ABSTRACT
Ehlers–Danlos syndrome is a rare disease and a diagnostic challenge. This case report serves to remind the clinician that it is important to identify all affected patients in order to prevent complications.
LEARNING POINTS
- Ehlers–Danlos syndrome is a rare disease of connective tissues that is often underdiagnosed.
- Late diagnosis leads to chronic pain and complications, limiting daily activity.
- Examination of all family members is essential when a patient is identified.
KEYWORDS
Ehlers–Danlos syndrome, rare disease, Beighton score, chronic pain, hypermobility, velvety skin
INTRODCTION
A 36-year-old woman was admitted to an internal medicine unit with hypertensive crisis associated with generalized musculoskeletal pain. She was a current smoker, cannabis user, regularly drank alcohol and had untreated hypertension. Some years before, she had suffered major trauma in a car accident and underwent multiple surgical procedures. She was taking hormonal therapy for polycystic ovarian syndrome and benzodiazepines for a chronic anxiety state. In order to identify the cause of her hypertension, she underwent several medical evaluations that found slight hyperaldosteronism with hypokalaemia. She was, therefore, diagnosed as having secondary hypertension from the contraceptive pill and use of illegal drugs.
The patient also complained of chronic intensive pain affecting her back, wrists, shoulders, knees and hips that was physically and psychologically disabling and limited her activities of daily living. Her history revealed that she did not start to walk until she was 24 months old, and was always clumsy and had difficulty with her coordination. She had a family history of multiple joint dislocations, early osteoarthritis and recurrent epistaxis. The clinical examination showed generalized hypermobility with pronounced hypermobility in her peripheral joints, knees, shoulders and spine. She also had dorsal kyphosis, flat feet on standing and hallux valgus. Her skin was smooth and silky, with increased extensibility over the back of her hands. There was atrophic, white, enlarged ("cigarette paper") scarring on her arms and legs in areas of previous trauma (Fig. 1).
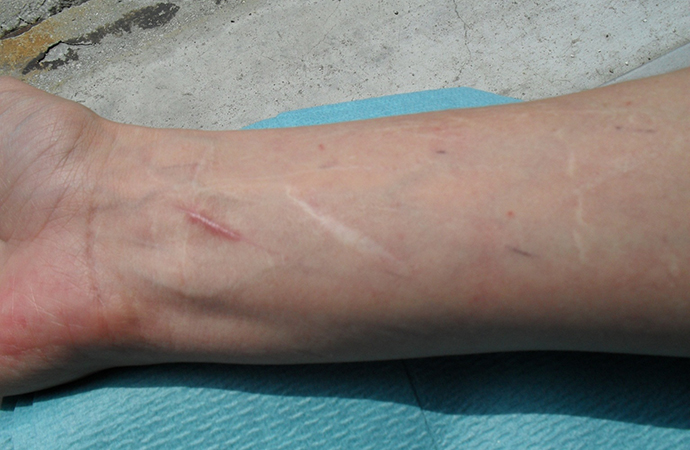
Figure 1 (click to enlarge)
Fig. 1 - Widened atrophic scarring and smooth and velvety skin, characterizing classical form EDS.
X-rays showed dystrophic cysts in the trochanteric region and cervical spondylo-arthrosis. Ocular anterior chamber abnormalities were detected. A thoracic CT evaluation revealed a slight dilatation of the proximal aortic root (approximately 3.6 cm) and a mild mitral regurgitation[1].
Orthopaedic consultation confirmed the presence of a joint hypermobility using the Beighton scoring system. A diagnosis of classical Ehlers–Danlos syndrome (EDS) type I–II was made on the grounds of clinical findings and her medical history.
The patient’s sister had been diagnosed with osteogenesis imperfecta based on a skin biopsy that showed an alteration of type I collagen. However, it is known that osteogenesis imperfecta is a rare genetic disorder caused by mutations in the genes coding for type I collagen and the most common symptoms are increased bone fragility and fractures in youth. Her sister had never complained of such symptoms. This could suggest that both patients are affected by classical type EDS, which can also be caused by an abnormality in type I collagen[2,3].
EDS is one of a large group of rare connective tissue disorders[4] and has the following major forms: classical (EDS type I–II), hypermobility (type III), vascular (type IV), kyphoscoliosis (type VI), arthrochalasia (type VIIA–VIIB) and dermatosparaxis (type VIIC)[5,6]. The prevalence of classical EDS has been estimated at 1:20,000. The molecular diagnosis of EDS is very time consuming and expensive and, therefore, is not used as a diagnostic tool. The diagnosis of the classic type can be established on clinical findings using the Villefranche criteria[7].
The three major criteria for classic EDS are as follows:
- skin hyperextensibility
- widened atrophic scarring signifying tissue fragility
- generalized joint hypermobility
Minor clinical criteria are as follows:
- smooth and velvety skin
- easy bruising
- molluscoid pseudotumours and subcutaneous spheroids
- muscle hypotonia
- delayed gross motor development
- dislocations/subluxations
- pes planus due to joint hypermobility
- manifestations of tissue extensibility and fragility (i.e. hiatal hernia, anal prolapse, mitral valve prolapse, cervical insufficiency and surgical complications such as postoperative hernias)
- a positive family history[8]
The disease has an autosomal dominant inheritance. Pain is very common[9]; it is nociceptive, responding to anti-inflammatory drugs, and neuropathic, responding to antidepressants, antiepileptics and opioids. When the diagnosis is made, patients should be subjected to screening for vascular, ligamentous and musculoskeletal abnormalities in order to control/prevent possible sequelae such as wound dehiscence and aortic or viscera rupture. As in many rare diseases, many patients remain undiagnosed.
In the classic form of the syndrome, there are mutations on the gene for collagen V, a fibrillar collagen co-expressed with type I collagen; this mutation produces a significant reduction of the protein in affected tissues[10]. On electron microscopy of the skin, the classic type shows a characteristic, but not diagnostic, "cauliflower" deformity of collagen fibrils[11]. Other genes such as the collagen I gene and tenascin gene have also been shown to be involved in the classic type of EDS[12-14]. About 90% of individuals with classic EDS have an identifiable mutation in COL5A1 or COL5A2, encoding the type V collagen, and, more rarely, an abnormal electrophoretic pattern for type I collagen caused by a mutation in the COL1A1 gene coding for it[15].
Although diagnosed late in life, our patient had all the clinical features required for diagnosis of classic EDS. She had the major criteria of elasticity of the skin at the back of her hands, white and baggy scars like cigarette paper, cervical kyphosis, a dorsal and lumbar hump, bilateral hallux valgus, hyperextension of the elbow >180°, shoulder instability and osteoarthritis of the knees. She also had the minor criteria of smooth and velvety skin, mild aortic bulbar ectasia, mild mitral valve insufficiency, tortuosity of the distal part of the right internal carotid artery and accentuation of the ocular papillary excavation. Moreover, her sister suffers from a collagen type I disorder, suggesting that they both have a rare form of classic type EDS involving the COL1A1 gene, which is associated with more joint involvement than skin alterations.