ABSTRACT
Henoch-Schönlein purpura is an IgA-mediated immune vasculitis which is characterized by purpuric lesions and osteoarticular, intestinal and sometimes renal manifestations. The histopathological substrate of this entity is leucocytoclastic vasculitis (LCV) with IgA deposits seen on immunohistochemistry. We here report the case of a 27-year-old woman with abdominal pain and cutaneous purpura. Upper and lower endoscopic exploration showed purpuric lesions in the rectum but not in the stomach. Skin biopsy revealed LCV. IgA deposits were seen only in gastric mucosa. The patient was treated with corticoids which led to improvement of both the cutaneous and digestive symptoms. This case suggests that gastrointestinal biopsies of both normal and abnormal mucosa should be taken in Henoch-Schönlein purpura, especially in patients with atypical forms.
LEARNING POINTS
- The diagnosis of Henoch–Schönlein purpura may be difficult, especially in patients with atypical forms.
- Identification of IgA deposits is important for the diagnosis; these deposits may be absent in skin biopsies but present in gastrointestinal mucosa despite the absence of lesions on endoscopy.
- Therefore, taking gastrointestinal biopsies of both involved and uninvolved mucosa is important.
KEYWORDS
Henoch–Schönlein purpura, gastrointestinal disease, endoscopy
INTRODUCTION
Henoch–Schönlein purpura (HSP) is an acute small-vessel leucocytoclastic vasculitis (LCV) that affects both infants and adults, although it is most common in childhood [1]. The pathogenesis of HSP is not well understood, but IgA is known to have a pivotal role [1, 2].
According to EULAR criteria [3], the diagnosis of HSP is based on the mandatory presence of purpura or petechiae with lower limb predominance plus one of the following four criteria: (i) abdominal pain; (ii) IgA seen on histopathology; (iii) arthritis or arthralgia; (iv) and renal involvement.
We here report a case of HSP with cutaneous and gastrointestinal (GI) manifestations where the diagnosis was supported by the presence of IgA in normal gastric mucosa.
CASE REPORT
A 27-year-old woman was hospitalized because of abdominal pain and cutaneous purpura on her upper and lower extremities. Laboratory tests indicated an inflammatory syndrome. A skin biopsy revealed LCV.
During her hospital course, the patient complained of abdominal pain and diarrhoea. Upper GI endoscopic examination did not reveal any abnormalities. However, rectosigmoidoscopy showed numerous purpuric lesions in the rectum (Fig. 1) and immunohistochemical examination revealed IgA deposits in the vascular walls of the gastric mucosa. This test was negative in both skin and rectal biopsies.
Consequently, the patient was diagnosed with Henoch–Schönlein purpura and was treated with corticotherapy. This resulted in resolution of the skin eruption and digestive symptoms.
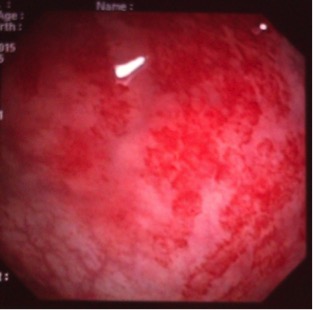
Figure 1 (click to enlarge)
Figure 1. Rectosigmoidoscopy showing purpuric lesions in the rectum
DISCUSSION
HSP is an IgA-associated small-vessel LCV[2]that occurs commonly in children[1, 4]. It is characterized by non-thrombocytopenic palpable purpura, arthralgia/arthritis, bowel angina and haematuria/proteinuria[1, 4]. The diagnosis of HSP is based on the 2010 EULAR criteria[3]. According to these guidelines, a patient is classified as having HSP in the presence (mandatory) of purpura or petechiae with lower limb predominance plus one of the following four criteria: (i) abdominal pain; (ii) IgA seen on histopathology; (iii) arthritis or arthralgia; or (iv) renal involvement.
GI involvement occurs in 50–75% of patients[1] and includes acute abdominal pain, nausea, vomiting, bloody stools and upper GI haemorrhage[4]. HSP can present with severe GI involvement which can be life-threatening in the short term[4].
Endoscopic examination is very important for detecting the GI manifestations of HSP. A large biopsy of purpuric lesions can detect the vasculitis in the small vessels of the mucosa. Direct immunofluorescence of tissue specimens from the skin, GI tract or kidney may show IgA and C3 deposits in both involved and uninvolved tissue[1]. In adults, the diagnosis of HSP is more difficult, especially in atypical forms. Many other diagnoses involving LCV must be ruled out, including infection, drug effects, malignancy and connective tissue disease[5].
According to the EULAR recommendations[3], in the case of atypical forms (atypical cutaneous manifestations or incomplete forms of disease), the presence of IgA deposits (which has a sensitivity and specificity of >80%) is required. The biopsy site is not specified in the guidelines. However, HSP is thought to be a multi-organ vasculitis involving the skin, joints, GI tract and kidneys, and so histological lesions can be identified in skin, renal and GI biopsies.
In this case report, IgA deposits were only present in normal gastric mucosa despite the typical cutaneous manifestations and the rectal lesions. Therefore, systematic GI biopsies of both abnormal and normal mucosa may be helpful for the diagnosis in selected patients presenting with atypical presentations, especially as it seems to be safer to take a GI biopsy than a renal biopsy.
Although evolution may be spontaneously favourable, numerous observations have suggested digestive manifestations may resolve with corticosteroids[4], as reported in this case.
CONCLUSION
As the presence of IgA deposits in the vessel wall is an important marker for the diagnosis of HSP, we suggest systematically taking GI biopsies from both involved and uninvolved mucosa, especially in atypical forms.