ABSTRACT
Interrupted aortic arch (IAA) is a rare congenital abnormality with only a few cases reported in adults. It is defined as complete loss of continuity between the ascending and descending portions of the aorta, and is usually associated with other cardiac defects. The diagnosis in adults should be suspected in the presence of refractory hypertension, a careful physical examination being crucial to early diagnosis. Magnetic resonance angiography (MRA) techniques can accurately characterize cardiovascular anatomy, and also provide information regarding heart chamber and valve function.
LEARNING POINTS
- Although rare, interrupted aortic arch (IAA) must be considered in the differential diagnosis of adults with refractory hypertension. A careful physical examination, with evaluation of femoral pulses and blood pressure in the four limbs, is essential for the diagnosis and for ensuring that the correct diagnosis is made promptly.
- This case highlights the value of magnetic resonance angiography (MRA) in radiation-free non-invasive evaluation of adults with congenital aortic abnormalities, as it may provide clinically important haemodynamic information.
- Only a few cases have been reported in adults. We present an unusual case in which the diagnosis was not made until adulthood.
KEYWORDS
Interrupted aortic arch, refractory hypertension, bicuspid aortic valve, aortic coarctation
BACKGROUND
Interrupted aortic arch (IAA) is a rare congenital abnormality defined as a complete loss of anatomical continuity between the ascending and descending portions of the aorta. Up to 98% of cases are associated with additional cardiovascular anatomical defects[1].
Only few cases of IAA have been reported in adults[2]. Survival into adulthood relies on the development of an extensive arterial collateral network, which is essential for the maintenance of distal blood flow. The diagnosis in adults should be suspected in the presence of hypertension, diminished or absent femoral pulses, and a differential between upper and lower limb blood pressure.
The authors describe an unusual case of a 48-year-old female patient in whom the diagnosis of IAA was not made until adulthood and which was successfully corrected surgically.
CASE REPORT
A 48-year-old female patient with a history of refractory hypertension since an early age and active smoking was admitted to the emergency room with hypertensive acute pulmonary oedema, referred as sudden dyspnoea and oppressive pre-cordial pain of about 2 hours duration.
On physical examination, the patient had shortness of breath, a respiratory rate of over 30 breaths per minute, sudoresis, and jugular venous distention at 45º. Systolic blood pressure was up to 200 mmHg and diastolic up to 100 mmHg in both upper limbs. The patient was tachycardic with a heart rate of 132 bpm, and showed peripheral oxygen saturation (in room air) of 78%.
Cardiac auscultation revealed rhythmic, hypophonetic cardiac sounds, while pulmonary auscultation revealed scattered crackles in both lungs, with no peripheral oedema. The 12-lead electrocardiogram showed sinus tachycardia and a left bundle branch block. Arterial blood gas analysis revealed uncompensated metabolic acidaemia (pH 6.9, pCO2 54.5 mmHg, HCO3− 16.9 mmol/l) with hyperlacticaemia (11.3 mmol/l). Admission laboratory studies showed no significant changes, including myocardial necrosis biomarkers. Transthoracic echocardiography in the emergency room revealed left chamber dilation, aortic root ectasia, a bicuspid aortic valve, global hypokinesia, and moderate depression of left ventricular systolic function.
Due to the lack of response to medical therapy, orotracheal intubation and ventilatory support were necessary, and the patient was admitted to the cardiac intensive care unit. Emergent cardiac catheterization excluded coronary disease.
During hospitalization, the patient showed a favourable clinical evolution, allowing extubation in the first 12 hours, and progressive improvement of the pulmonary congestion. However, she remained hypertensive, requiring quadruple therapy. On physical examination, blood pressure between arms and legs differed by up to 40 mmHg, with no perceptible delay on femoral pulses. The patient did not have any features of diGeorge syndrome.
Echocardiogram was repeated and confirmed severe left ventricular and mild left atrial dilatation, a bicuspid aortic valve with mild grade insufficiency, and global left ventricular hypokinesia with severely depressed global systolic function (ejection fraction 32%) (Fig. 1).
The presence of refractory hypertension, a blood pressure differential, subtle bilateral rib notching and a bicuspid aortic valve alerted us to the possibility of IAA. Magnetic resonance angiography (MRA) was performed and reveal dilated hypertensive cardiopathy with moderate depression of systolic function and severe coarctation/aortic interruption at the isthmus, with significant collateralization through the internal mammary arteries, the upper and lower epigastric arteries, and the intercostal arteries, which appeared diffusely dilated (Fig. 2).
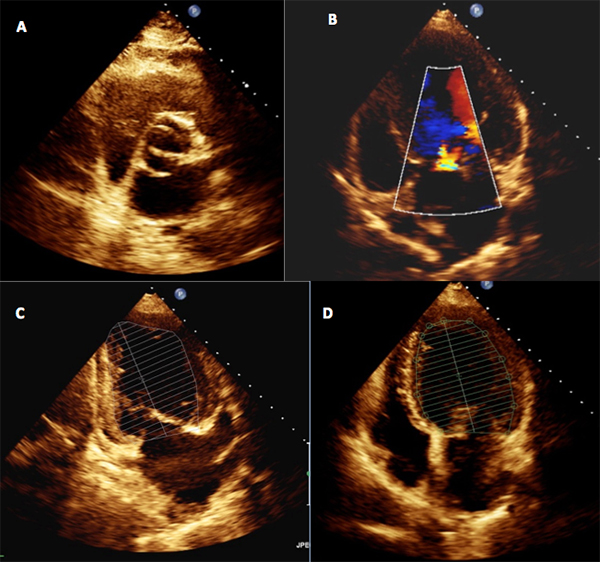
Figure 1 (click to enlarge)
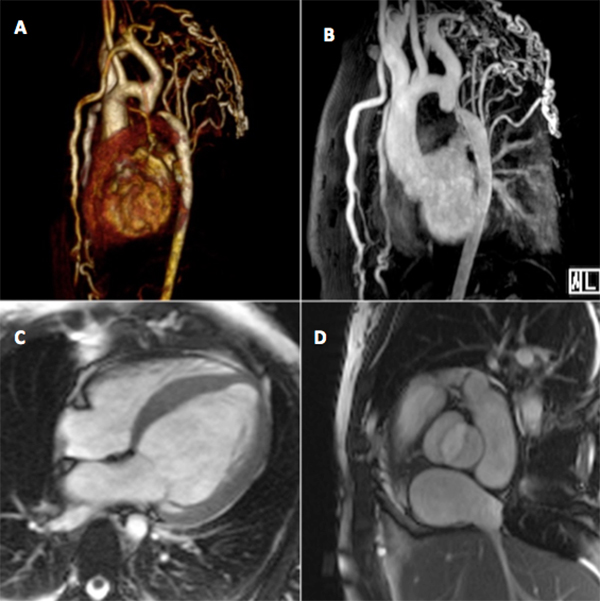
Figure 2 (click to enlarge)
Figure 1. Transthoracic echocardiogram showing a bicuspid aortic valve (A) with mild grade insufficiency (B), and severe left ventricular (LV) dilatation (median LV volume 103 ml/m2) and global LV hypokinesia with severely depressed global systolic function (ejection fraction 32%) and preserved right ventricle function (C and D)
Figure 2. (A) Three-dimensional volume rendered (3D-VRT) and (B) maximal intensity projection (MIP) images of the thoracic aorta reconstructed from contrast-enhanced magnetic resonance angiography showing aortic arch interruption and multiple dilated intercostal, internal mammary and subclavian arteries, consistent with extensive aortic-systemic collateralization. Cine steady-state free precession (SSFP) images in (C) 4-chamber and (D) aortic root views show concentric left ventricular hypertrophy and a bicuspid aortic valve
Computed tomography angiography (CTA) revealed a bicuspid aortic valve and about 2 cm after the origin of the left subclavian artery, complete discontinuity of the aortic lumen, with a distance of 4 mm between the proximal and distal tops (Fig. 3). Surgical correction was performed with interposition of a 16 mm Dacron graft. The patient recovered uneventfully with no complications. CTA performed 9 months later demonstrated a successful result, with a permeable graft, without significant stenosis or images suggesting leaks (Fig. 4).
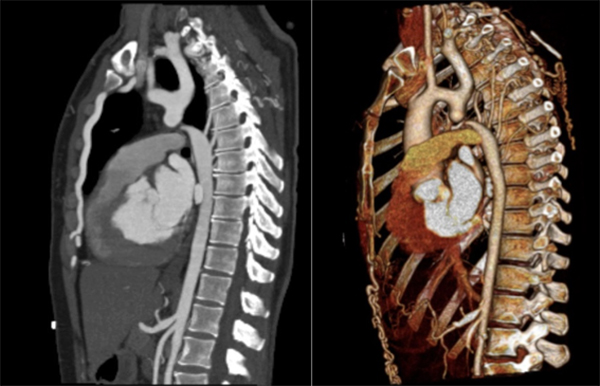
Figure 3 (click to enlarge)
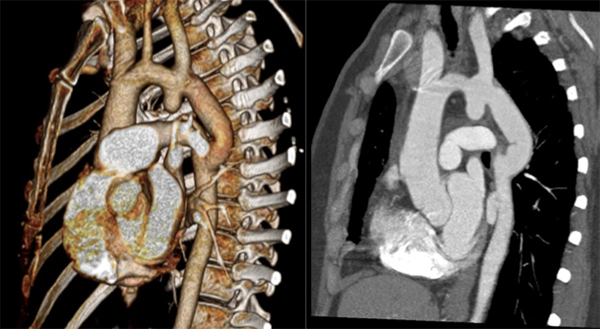
Figure 4 (click to enlarge)
Figure 3. Computed tomography angiogram showing a type A interrupted aortic arch, with complete discontinuity of the aortic lumen distal to the origin of the left subclavian artery
Figure 4. Computed tomography angiogram showing the permeable graft 9 months after surgical correction
DISCUSSION
IAA is a rare congenital malformation accounting for less than 1% of all cases of congenital heart disease[2]. It is an extreme form of aortic coarctation and is defined as a complete loss of luminal and anatomical continuity between the ascending and descending portions of the aorta. Steidele first described it in 1778, and Celoria and Patton introduced the first classification system in 1959, which is still used[3].
In this classification, IAA is divided in three different types according to the site of the aortic discontinuity: type A is distal to the origin of the left subclavian artery (LSA), type B is between the left common carotid artery (LCCA) and the LSA, and type C between the innominate artery and the LCCA[4]. Type B is the most frequent in neonates (53%), and is associated with DiGeorge syndrome and chromosome 22q11.2 deletion[4], while type A is more frequently reported in adults (79%)[2].
Over 98% of patients with IAA have other congenital cardiac anomalies such as patent ductus arteriosus, truncus arteriosus, ventricular septal defect, bicuspid aortic valve, left ventricular outflow tract obstruction, aberrant innominate arteries and aortopulmonary window[3]. A bicuspid aortic valve occurs in one third to one half of patients.
Almost all cases are diagnosed in the neonatal period and present with severe congestive heart failure with rapid clinical deterioration. If untreated, 90% of affected infants die in the first year of life, the majority in the first few days[5].
The presentation in the few reports in adults differs markedly from that in children, and ranges from lack of symptoms to refractory hypertension, headache, claudication, malaise, differential blood pressure between the arms and legs, and congestive heart failure. About 13% of patients have life-threatening complications at presentation[2].
Survival into adulthood relies on the development of an extensive collateral network, which is essential for the maintenance of distal flow. The presence of a ductus arteriosus that closes gradually during childhood is crucial to the development of this collateral network.
The differences described above between children and adults support the theory that in some cases the initial defect is a coarctation that evolves to complete obstruction of the lumen[6]. In fact, type A IAA and severe coarctation cases may show clinical and anatomic similarities[7], with aortic coarctation being much more frequent, representing 4–6% of all congenital cardiac defects[8]. In this case, the morphology and location of the obstruction distal to the left subclavian artery raises the possibility that an undiagnosed aortic coarctation may have evolved to complete occlusion.
The diagnosis in adults should be considered in the presence of refractory hypertension. Physical examination is crucial to detect murmurs due to the extensive collateral circulation, a decrease or delay in the femoral pulses, and the upper to lower limbs blood pressure differential.
Chest x-ray may show cardiomegaly and/or a notch in the posterior ribs. Echocardiography, despite some limitations evaluating the aortic arch and descending aorta, is the technique of choice for the detection of other cardiac changes. CTA and MRA are useful complementary imaging methods[5]. Because of its ability to accurately define aortic and cardiac anatomy, MRI can be used to identify the site and length of the interruption, the origin of the great vessel and the associated congenital cardiac defects, and may be particularly beneficial in the work-up of suspected IAA when the defect cannot be adequately evaluated by an echocardiogram[9].
Depending on the anatomy and the location of the interruption, there are different surgical options for correction. In adults, the most frequent surgical approach is a single intervention with an extra-anatomical conduit[10]. Percutaneous stenting, although challenging, is feasible and an effective alternative to surgery.
Surgery is successful in most patients, showing low postoperative mortality and improvement or even resolution of the complaints of hypertension and claudication.
Patients who refuse or who are not suitable for surgery/stenting should be closely followed up and receive optimized medical therapy.