ABSTRACT
A 72-year-old woman presented with a complex partial seizure and right hemiparesis, after a four-week history of cognitive decline, apraxia and speech disturbance. She previously had chronic lymphocytic leukaemia (CLL) and had finished chemotherapy one year prior to presentation. She was receiving monthly intravenous immunoglobulins for bronchiectasis. Brain imaging showed hypodensity in the left temporo-parietal regions. Cerebrospinal fluid was positive for the JC virus, leading to a diagnosis of progressive multifocal leucoencephalopathy (PML). She remains alive, eight months following initial presentation. The case was valuable for reflective practice in avoiding diagnostic (confirmation) bias because the treating team pursued an incorrect diagnosis of stroke and secondary seizure after radiology findings appeared consistent with this. Additionally, PML has not previously been reported in individuals with CLL receiving immunoglobulin therapy, and may explain the relatively benign course in this individual patient. This offers a potential research question for disease modifying treatments in PML.
LEARNING POINTS
- This case highlights new insights into an uncommon but important condition: always consider progressive multifocal leucoencephalopathy when immunocompromised patients present with neurological symptoms.
- A full differential diagnosis should always be considered, even in the context of a more ‘plausible’ diagnosis.
- Avoid premature closure and confirmation bias as cognitive errors in diagnostic reasoning.
KEYWORDS
Chronic lymphocytic leukaemia, progressive multifocal leucoencephalopathy, immunocompromised
CASE PRESENTATION
A 72-year-old right-handed woman presented to the Emergency Department with a thirty-minute episode of right arm and leg shaking, right facial droop and expressive dysphasia. She remained conscious throughout the episode, but was incontinent of urine. The paramedics noted a right hemiparesis. The patient’s husband reported a four-week history of significant cognitive change, e.g. misplacing her belongings, forgetting how to use household appliances and difficulty performing simple tasks, such as pouring water into a cup.
She had a past medical history of chronic lymphocytic leukaemia (CLL), which was diagnosed four years prior to presentation. She had received the ‘FCR-lite’ chemotherapy regimen (fludarabine, cyclophosphamide and rituximab) with curative intent. The treatment course finished one year prior to presentation. She was receiving monthly intravenous immunoglobulin for a diagnosis of bronchiectasis and acquired hypogammaglobulinaemia post-chemotherapy. Her medications were aspirin, bisoprolol and an inhaler.
On arrival, she had right facial weakness, with increased tone in her right arm and leg, with 4/5 power. Reflexes were present, with an upgoing right plantar reflex. She had severe expressive and receptive dysphasia. There was past pointing, dysdiadochokinesis and sensory neglect on the right.
Blood tests revealed a mild anaemia (Haemoglobin level 119 g/dL), but were otherwise unremarkable.
CT brain scan (Fig. 1a) showed an area of hypodensity in the left temporo-parietal region, which appeared to be consistent with an old infarction. MRI with diffusion-weighted imaging showed hypodensity in the same region on T1-weight images (Fig. 1b), and hyperdensity on T2-weighted images (Fig. 1c), but with no diffusion restriction. Appearances were felt to represent a previous infarction.
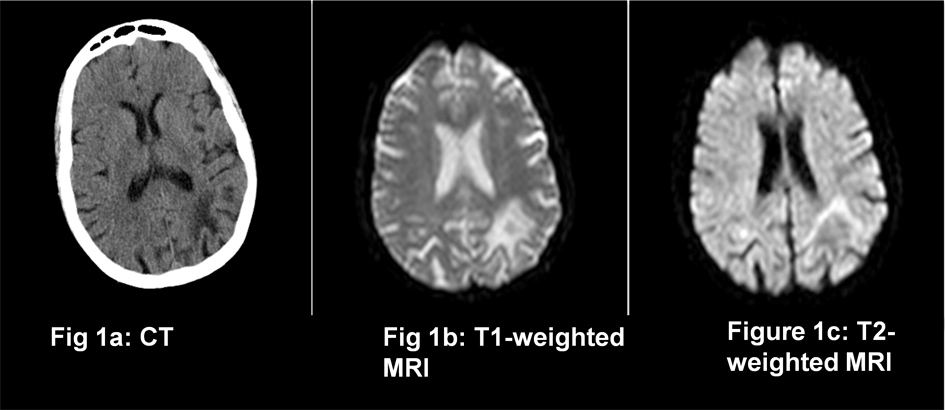
Figure 1 (click to enlarge)
Figure 1. CT (1a) and MRI (1b and 1c) findings on presentation
Differential diagnosis
The history is of a focal seizure with short preceding history of focal neurological symptoms and cognitive decline. MRI appearances were reported as being consistent with mature ischaemic areas, leading to a working diagnosis of recent stroke and secondary seizure. The patient was treated with antiplatelet therapy.
Given the diagnosis of CLL an important differential diagnosis is brain involvement of Richter’s syndrome (the transformation of CLL into a high grade B cell lymphoma). Additionally, progressive focal neurological symptoms in an immunocompromised patient could represent Progressive Multifocal Leucoencephalopathy.
Outcome and follow up
Lumbar puncture revealed an acellular cerebrospinal fluid with normal protein and glucose levels. Several weeks later, viral studies revealed 774 copies/ml of the JC virus.
She was stable as an in-patient, with improvement after physiotherapy. She was discharged home with palliative care. She had a further hospital admission one month later with pneumonia, and was treated supportively. However, eight months following her initial presentation, she remained alive and at home.
Diagnosis
Progressive multifocal encephalopathy (PML) secondary to JC virus, one year post chemotherapy for CLL.
DISCUSSION
PML is a progressive, demyelinating disorder that affects brain oligodendrocytes of immunocompromised individuals. The infectious agent is the JC virus (named after John Cunningham, the first patient diagnosed with the condition in 1958)[1-4]. Asymptomatic infection with JC virus occurs early in life and is common in healthy individuals. The virus then becomes latent and persists in the kidneys, lymphoid tissue and leukocytes. During periods of immune system dysregulation, the virus reactivates and travels to brain oligodendrocytes and astrocytes, where it induces cytolysis, resulting in cell death and demyelination[2,4].
It is uncommon, and was initially reported most during the AIDS pandemic, only rarely being associated with lymphoproliferative disorders. More recently, it has been associated with individuals receiving fludarabine and rituximab therapy. Additionally, it is now well recognised in patients with multiple sclerosis receiving natalizumab[2,4,5] Demyelinating lesions (which can be focal or multifocal) develop in the frontal or temporo-parietal areas and clinical features relate to the area of brain affected. Common presenting features include speech, motor and visual disturbance, cognitive impairment, and apraxia. Diagnosis is made based on clinical features, magnetic resonance imaging (classically showing hypointense lesions on T1-weighted images and hyperintensity in T2-weighted images without evidence of acute diffusion restriction) and CSF PCR (Polymerase Chain Reaction). Sensitivity of PCR is variable, so repeat testing should occur if clinical suspicion persists; alternatively brain biopsy should be considered[1,2,4]. Treatment involves immune reconstitution where possible (such as anti-retroviral therapy in HIV patients, and plasma exchange in natalizumab recipients), but the prognosis is very poor and no disease modifying treatments have been proven to date[1,4].
This case was remarkable because the symptoms of PML developed a year after the chemotherapy course finished. Additionally, the patient was receiving immune reconstitution in the form of immunoglobulin therapy, which should be protective. To our knowledge, no patients have been reported to develop PML while receiving immunoglobulins in CLL. Immunoglobulin therapy has even been used as a treatment in natalizumab induced PML in multiple sclerosis, where it has been associated with an improvement in clinical state[6]. This could explain the fact that the patient was alive and living at home eight months following presentation, when the prognosis in patients with haematological malignancy is generally very poor[7]. As there are no disease-modifying agents to date for this condition, this could be an important area for future research.
CONCLUSIONS
Heightened awareness of this condition is important, particularly as biologic therapies are likely to play a more prominent role in the future.Although PML may be seen commonly in certain subspecialties, it is a rare presentation to the internist. Thus, PML should be actively considered and excluded in immunocompromised patients presenting with progressive neurological symptoms. Rapid diagnosis leads to more effective counselling and prognostication, entry into clinical trials, and advance care planning. The diagnosis was initially delayed, with treatment and investigation more focused towards the management of stroke and secondary seizure – a more common diagnosis on the internal medicine ward. This is an example of cognitive errors in diagnostic reasoning: premature closure and confirmation bias when relying on the imaging findings. Finally, the case poses an important area for future research in disease-modifying therapies in PML.