ABSTRACT
Light-chain deposition disease is a rare paraproteinaemia characterized by deposition of monoclonal light-chain immunoglobulins with a non-amyloid structure, most frequently affecting the kidney. The authors present the case of a 58-year-old man admitted due to haemorrhagic shock caused by spontaneous splenic rupture. Investigation showed coagulopathy, homogeneous hepatomegaly with elevated cholestasis enzymes but normal bilirubinaemia, and a nephrotic syndrome with monoclonal kappa light chains. Liver and bone marrow biopsies revealed light-chain deposition disease. Splenic rupture as the presentation of a paraproteinaemia is rare. The diagnosis of light-chain deposition disease should be considered in case of a monoclonal plasma cell disorder with Congo-red negative deposits on histology.
LEARNING POINTS
- Spontaneous splenic rupture is a rare presentation of a paraproteinaemia.
- Light-chain deposition disease is a rare disease with non-fibrillar deposition of monoclonal light-chain immunoglobulins most frequently affecting the kidney.
- Light-chain deposition disease should be suspected in case of a monoclonal plasma cell disorder with Congo-red negative deposits on histology.
KEYWORDS
Light-chain deposition disease, paraproteinemia, splenic rupture
INTRODUCTION
Light-chain deposition disease (LCDD), heavy-chain deposition disease, and light- and heavy-chain deposition disease are a rare group of paraproteinaemias characterized by the deposition of monoclonal immunoglobulins with a non-fibrillar structure and hence Congo-red negative deposits[1,2]. The authors describe the diagnostic pathway of a 58-year-old man diagnosed with LCDD following splenic rupture.
CASE DESCRIPTION
A 58-year-old man presented to the emergency department with a 1-day history of left back pain that evolved to generalized abdominal pain, coffee-ground vomiting and prostration. There was no recent trauma. The patient was a smoker, with a previous peptic ulcer, was medicated with bromazepam for insomnia, and had consumed 50 g of alcohol daily until 6 months previously.
On admission, he was hypotensive (89/56 mmHg), tachycardic (99 bpm), pale and with pain in the upper quadrants on abdominal palpation, with guarding. A peptic ulcer was initially suspected, but abdominal radiography did not show free gas. The nasogastric tube drainage fluid consisted of gastric contents with a small amount of digested blood. A laboratory investigation (Table 1) showed lactic acidosis, acute anaemia, coagulopathy, acute kidney failure with hyperkalaemia, and elevated cholestasis enzymes. Abdominal CT (Fig. 1) revealed splenic rupture with possible splenic infarction, an exuberant haemoperitoneum and homogeneous hepatomegaly measuring 17 cm. The haemorrhagic shock due to spontaneous splenic rupture required an emergent total splenectomy.
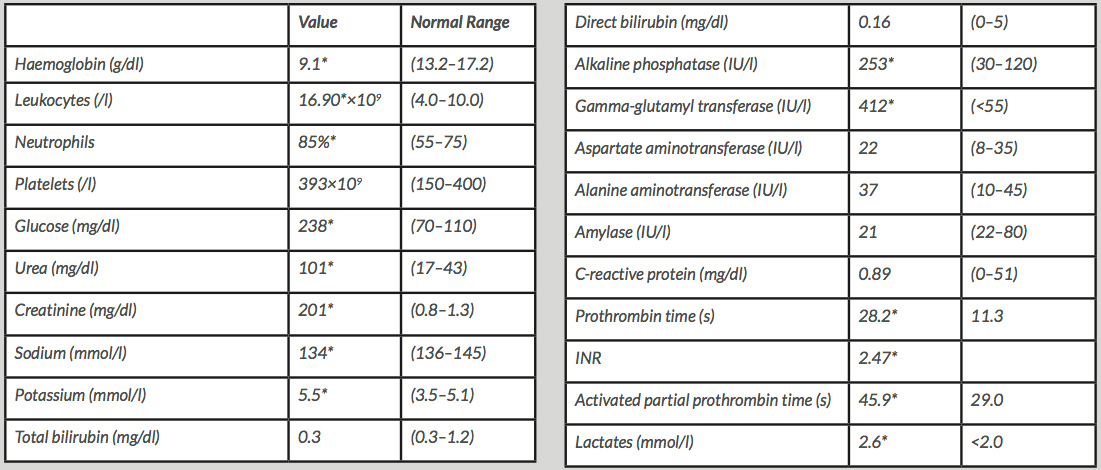
Table 1 (click to enlarge)
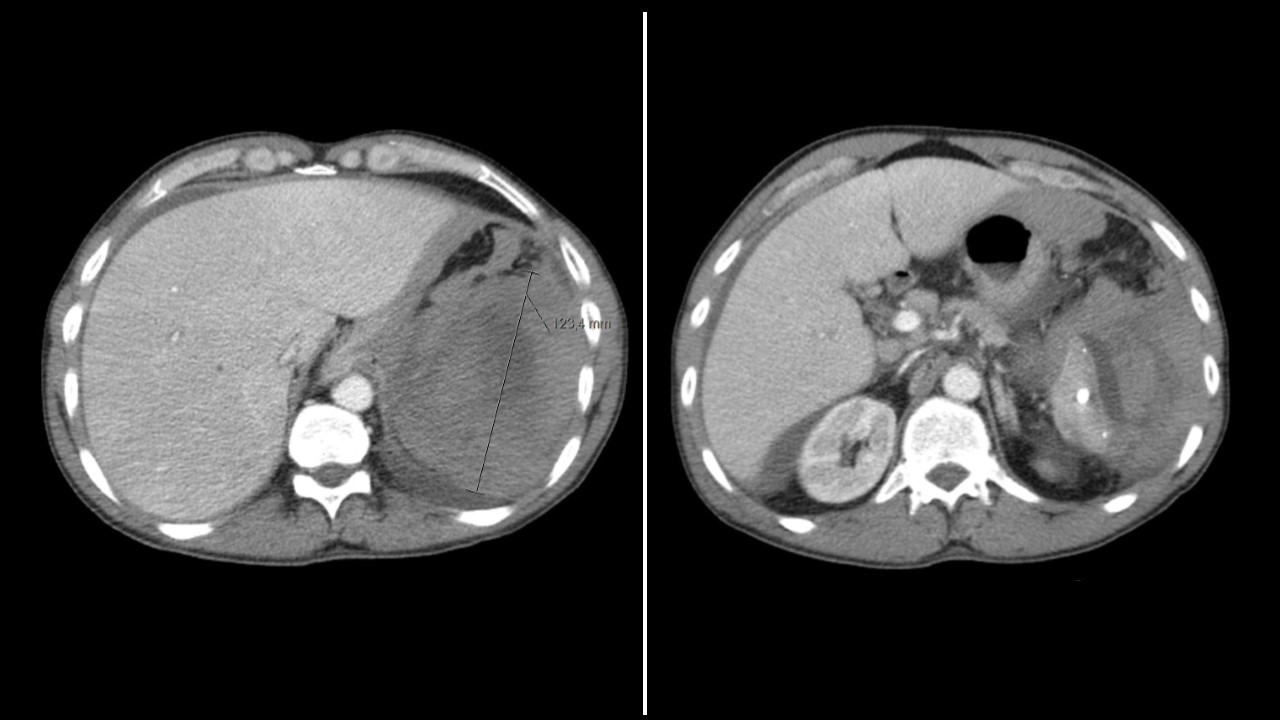
Figure 1 (click to enlarge)
Table 1. Laboratory investigation results *Value outside the normal range
Figure 1. Abdominal computerized tomography: Splenic rupture with an area of possible splenic infarction, large subcapsular splenic hematoma, exuberant hemoperitoneum and homogenous hepatomegaly of 17cm
The patient had a good clinical evolution in the post-operative period except for worsening of the coagulopathy (PT 31.8 s, INR 2.78 and aPTT 53.9 s) with no bleeding, and elevation of cholestasis enzymes (alkaline phosphatase 535 IU/l and gamma-glutamyl transferase 418 IU/l).
The initial work-up did not reveal the aetiology of the liver disease, but did exclude viral hepatitis, haemochromatosis, Wilson’s disease, alpha1-antitrypsin deficiency and drug consumption. A normal platelet count, transaminases and bilirubinaemia made alcoholic liver disease less likely, despite the previous alcohol consumption. However, there was marked hypoproteinaemia (4.1 g/dl) with hypoalbuminaemia (2.1 g/dl), dyslipidaemia (total cholesterol 270 mg/dl) and hypogammaglobulinaemia on serum protein electrophoresis. Despite the bleeding, fibrinogen was normal, ruling out consumption coagulopathy. An abdominal ultrasound showed homogeneous hepatomegaly and a normal biliary tract and excluded hepatic vascular complications. In a broader investigation, an immunological study was negative but urinalysis revealed massive proteinuria. There were 9.8 g of protein in the 24-hour urine and so a nephrotic syndrome was diagnosed (nephrotic proteinuria, dyslipidaemia, hypoalbuminaemia and probable splenic thrombosis). The lack of significant oedema was explained by treatment with furosemide, prescribed in the presumed context of excessive intravenous fluids. An infiltrative disease was suspected because of the pattern of liver disease with nephrotic syndrome.
Magnetic resonance cholangiopancreatography exhibited slight peribiliary oedema, while stool examination for parasites was negative, as was alpha-fetoprotein. Regarding the nephrotic syndrome, despite the hypogammaglobulinaemia, serum immunofixation revealed monoclonal IgA kappa, monoclonal kappa light chains were detected in immunofixed urinary protein, and the serum free light-chain ratio was abnormal (9.06). Serum angiotensin-converting enzyme, serum calcium and 24-hour calciuria were normal and a more detailed immunological study remained negative. To evaluate the coagulopathy, an aPTT mix 1:1 showed normalization of aPTT, indicating a factor deficiency, later identified as factor X deficiency (levels 0.03 U/ml, 4%).
The spleen histology which previously showed splenic phleboliths was revised but was negative for infiltrative disease and was Congo-red negative, as was an abdominal fat biopsy. The coagulopathy postponed the decision to carry out an organ biopsy but at this point, the patient was submitted to a transjugular liver biopsy with administration of prothrombin complex concentrate. Histology (Figs. 2 and 3) revealed acidophilic amorphous deposits in the stroma of hepatic lobules, with atypical Congo-red staining and birefringence but with immunohistochemistry positive for kappa light chains. An initial diagnosis of amyloidosis was assumed.
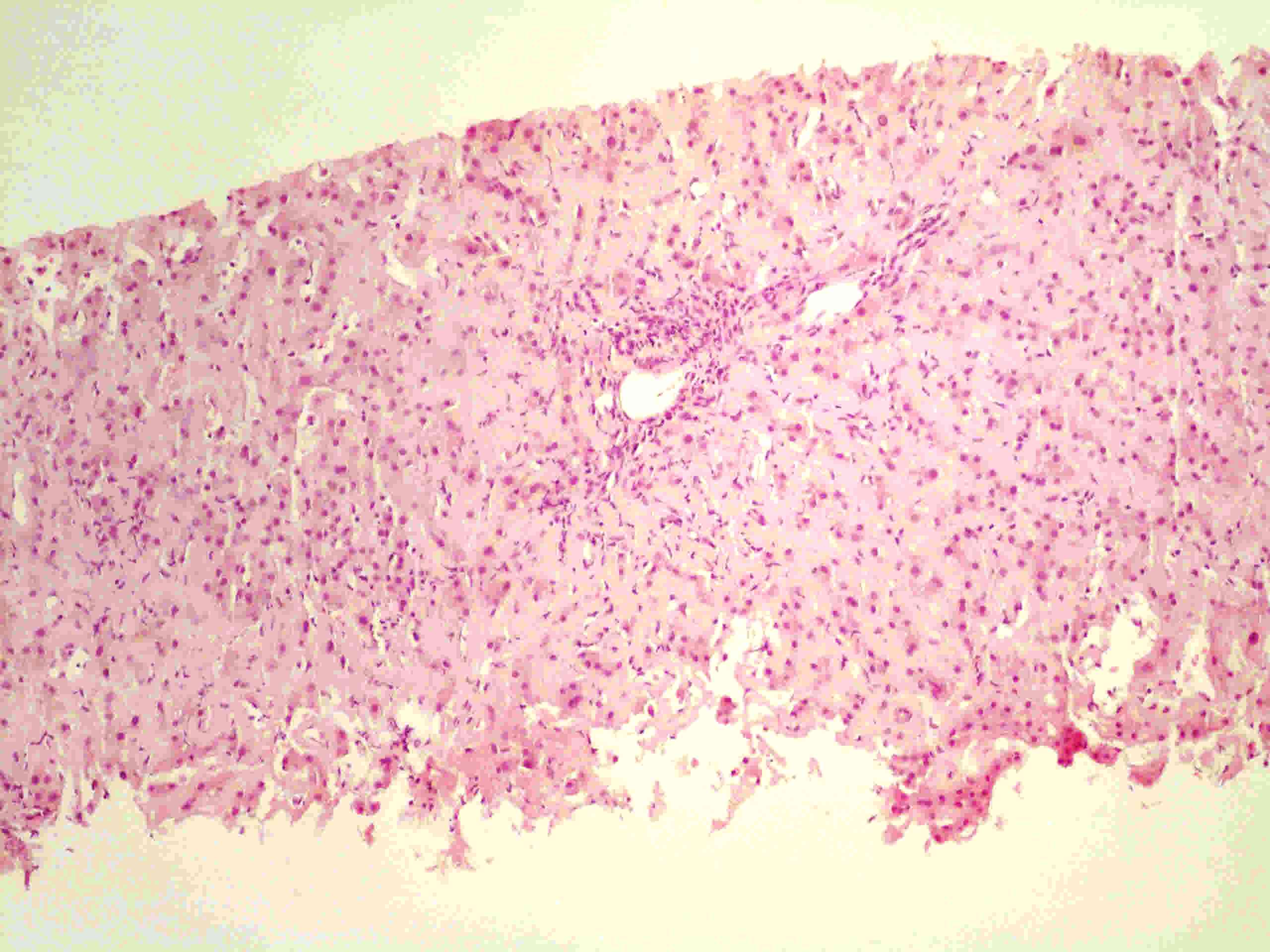
Figure 2 (click to enlarge)
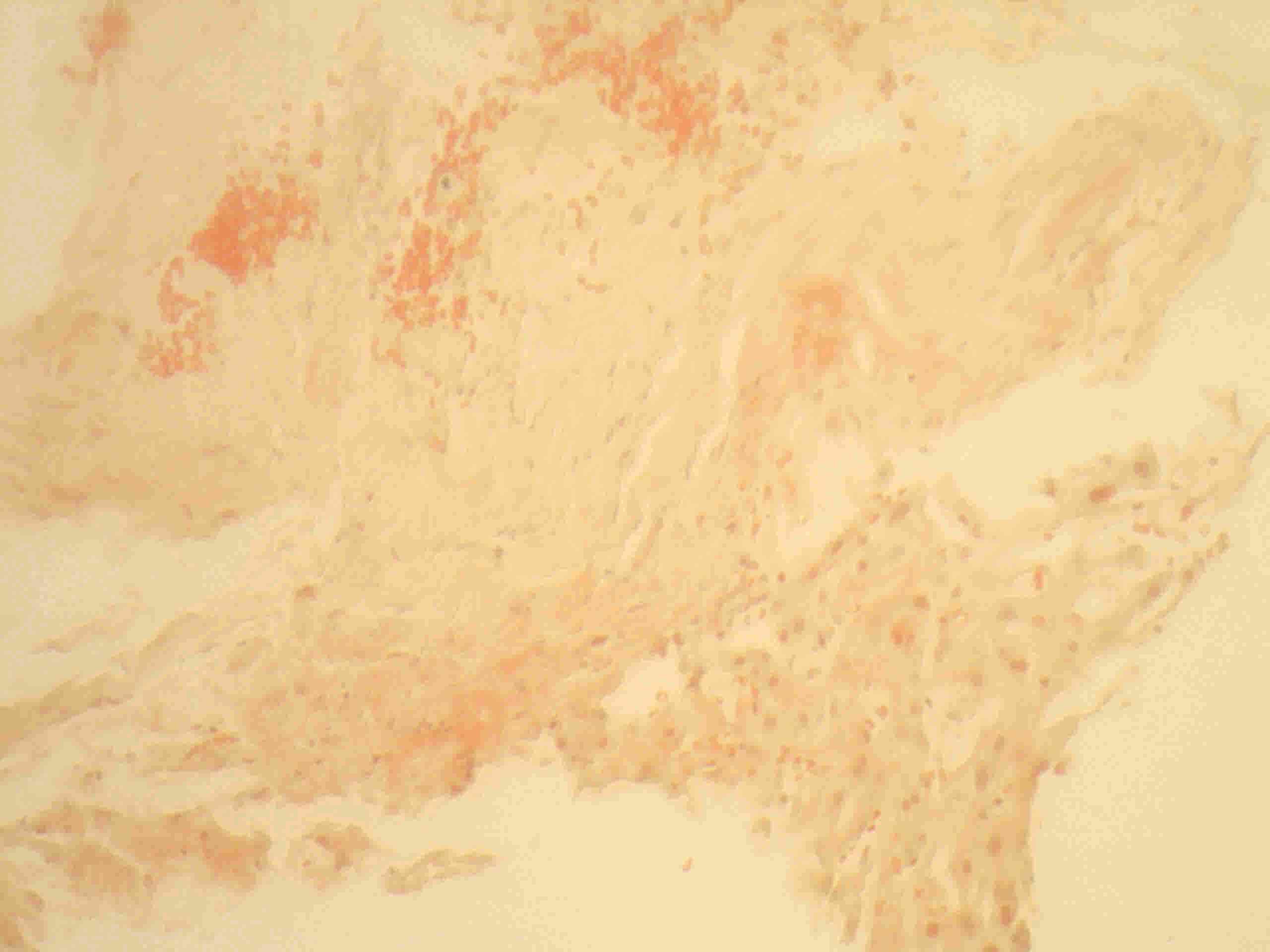
Figure 3 (click to enlarge)
Figure 2. Liver biopsy histology, Hematoxylin-eosin staining, 10x: Portal and peripostal inflammatory infiltrate and acidophilic amorphous deposits surrounding the hepatocytes
Figure 3. Liver biopsy histology, Congo-red staining, 20x: Amorphous deposits with atypical positivity
As part of the characterization of the amyloidosis, a bone marrow study showed 0.3% monoclonal kappa light-chain plasmocytes and infiltration of the bone marrow by an amorphous, hyaline material, negative for Congo-red staining but positive for kappa light chain on immunohistochemistry. Following histology review, light-chain deposition disease was diagnosed.
The patient started chemotherapy with cyclophosphamide, bortezomib and dexamethasone, but shortly afterwards developed progressive renal failure and is currently on dialysis.
DISCUSSION
SSpontaneous spleen rupture is a rare event in a paraproteinaemia, namely amyloidosis, and was the initial manifestation of amyloidosis in 71% of cases[3]. There seems to be an association with factor X deficiency[3]. Factor X deficiency is a rare coagulopathy that can be congenital or acquired[4]. Acquired factor X deficiency is usually due to vitamin K antagonists or a plasma cell dyscrasia, such as amyloidosis[4].
LCDD, a rare disease of non-fibrillar deposition of monoclonal light-chain immunoglobulins, usually manifests in the fifth or sixth decade of life with a male predominance, but the incidence is unknown[2]. In LCDD, the monoclonal light chains are of the kappa subtype in 92% of cases and it is typically associated with multiple myeloma or other lymphoproliferative disorders[2]. The kidney is the main affected organ, presenting with hypertension, kidney failure, proteinuria and haematuria with 62% of cases requiring dialysis[1,2]. However, involvement of other organs such as the heart, the liver and neural involvement, has also been described[2].
Without treatment, kidney failure will evolve to end-stage renal disease, but treatment options for LCDD are limited[1,5]. Steroids and melphalan, high-dose melphalan, autologous stem cell transplantation and, more recently, bortezomib-based chemotherapy, are some of the treatment options[5]. The median estimated length of survival for treated patients is 14 years[1].
The authors have described this case with an interesting diagnostic pathway to highlight spontaneous spleen rupture as a rare presentation of plasma cell disorders, and LCDD as a diagnosis to consider in Congo-red negative, amorphous deposits.