ABSTRACT
Behçet syndrome (BS) is a variable vessel vasculitis that has pleiotropic manifestations. A 43-year-old male with a previous diagnosis of Crohn’s disease (CD) presented with deep venous thrombosis and bilateral superficial femoral artery aneurysms. A diagnosis of BS was made, and the patient was treated aggressively with immunosuppressive therapy and bilateral bypass surgery, attaining a favourable outcome. CD has many features that overlap with BS, and it may be challenging to distinguish between these two conditions, as our case illustrates. Nonetheless, the combination of venous thrombosis and arterial aneurysms should point the clinician towards a diagnosis of BS.
LEARNING POINTS
- Behçet syndrome is a variable vessel vasculitis of unknown aetiology that has pleiotropic manifestations.
- Crohn’s disease has many overlapping features with Behçet syndrome, namely gastrointestinal, cutaneous, articular, ocular and cardiac manifestations.
- The combination of venous thrombosis and arterial aneurysms should point the clinician towards a diagnosis of Behçet syndrome.
KEYWORDS
Behçet syndrome, terminal ileitis, deep venous thrombosis, aneurysm, Crohn’s disease
CASE PRESENTATION
A 43-year-old Portuguese male was admitted to our hospital in 2017 for surgical correction of a left superficial femoral artery (SFA) aneurysm. The patient’s past medical history was remarkable for a diagnosis of Crohn’s disease (CD) nine years (2008) prior to the current presentation, in the context of oral ulcers, abdominal pain and bloody diarrhoea. At that time, a colonoscopy revealed isolated terminal ileitis with unspecific histological findings – mucosal and submucosal inflammatory infiltrate without granulomas or microabscesses. The laboratory tests were unremarkable. Sulfasalazine was commenced, and the abdominal pain and diarrhoea remitted. From 2008 to 2017, the patient continued to have occasional flare-ups of oral ulceration and was diagnosed with CD-related peripheral and axial spondyloarthritis in 2011 – treated with golimumab for 6 months (stopped due to self-reported intolerance) – and with myopericarditis in 2014 – treated with colchicine for 3 months. His current medication was sulfasalazine 1000 mg twice daily. There was no family history of autoimmune diseases, immunodeficiency or thrombosis.
One month before the present admission the patient presented to another hospital with a 5-day history of low-grade fever, asthenia, oral ulcers, and painful oedema of the left lower limb. Deep venous thrombosis of the left popliteal vein and bilateral SFA aneurysms were noted on the CT angiogram (Fig. 1). Rivaroxaban was commenced, and the patient was referred to our vascular surgery unit for further evaluation.
48 hours, followed by neurological recovery and progressive regression of the MRI lesions.
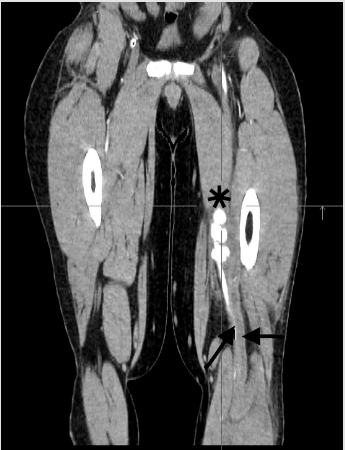
Figure 1 (click to enlarge)
Figure 1. CT angiogram of the lower limbs: coronal view illustrating the left superficial femoral artery aneurysm (black asterisk) and the left popliteal vein thrombosis (black arrows)
At our unit, physical examination revealed multiple oral ulcers, left lower limb oedema, and a pulsatile mass with an overlying bruit on the left thigh. Laboratory tests disclosed an inflammatory anaemia (haemoglobin 11 g/dL) and high acute phase reactants (C-reactive protein 5.4 mg/dL and erythrocyte sedimentation rate 48 mm/h); and were negative for HIV, hepatotropic viruses, HLA-B51, HLA-B27, cryoglobulins, rheumatoid factor, complement, the pathergy test, and antinuclear, anti-neutrophil cytoplasmic and antiphospholipid antibodies. Surgical correction of the left SFA aneurysm was immediately pursued, because it was significantly larger than the right SFA aneurysm and associated with a higher risk of rupture. Bypass surgery with aneurysm resection and a polytetrafluoroethylene interposition graft was performed, without complications. Histology of the aneurysm revealed leukocytoclastic vasculitis of the vasa vasorum (Fig. 2), with accompanying transmural infiltration of polymorphonuclear neutrophils and fibrinoid necrosis of the intimal layer.
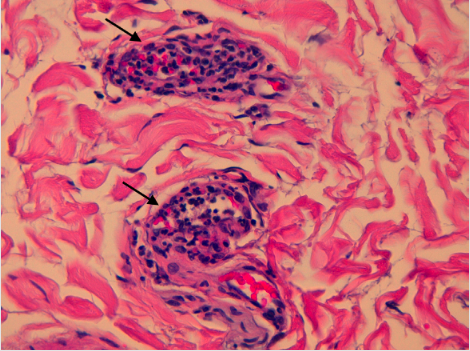
Figure 2 (click to enlarge)
Figure 2. Histology (haematoxylin–eosin at 400x magnification) of the aneurysm disclosing leukocytoclastic vasculitis of the vasa vasorum (black arrows)
Behçet syndrome (BS) with active mucosal and vascular involvement was diagnosed, and the patient was treated with 6-monthly intravenous pulses of cyclophosphamide 1 g, followed by azathioprine and bypass surgery of the contralateral SFA aneurysm. After 3 months of therapy, abnormal (>5x the upper limit of normal) liver function tests prompted the discontinuation of azathioprine. At 1 year follow-up, the patient was in remission, while on infliximab 5 mg/kg every 8 weeks, low-dose prednisolone (2.5 mg/day), and rivaroxaban 20 mg/day.
DISCUSSION
BS has an unknown aetiology, is classified as a variable vessel vasculitis, and usually presents in the third decade of life[1]. It has the highest incidence in countries located along the ancient Silk Road, stretching from Asia to the Mediterranean countries, but is not confined to these geographical areas, as our case illustrates.
BS typically runs an unpredictable course, with periods of recurrence and remission. It generally starts with a single manifestation (often oral ulcers) and, as the disease progresses, other manifestations eventually appear[2]. Besides oral ulceration, clinicians can find genital ulcers, cutaneous lesions (e.g. pseudofolliculitis, panniculitis), ocular disease (e.g. non-granulomatous panuveitis), arthralgias and arthritis (often nonerosive), neurological manifestations, venous thrombosis (e.g. lower extremities, inferior vena cava, cerebral venous sinuses), arterial aneurysms (typically of the pulmonary, abdominal aorta or peripheral arteries), gastrointestinal involvement, and cardiac disease (e.g. pericarditis, myocarditis), among others[1,2]. Among these, vascular complications are present in up to 40% of cases – 75% of which are venous in nature, whereas the other 25% are arterial – and associated with potentially life-threatening complications[3].
As our case illustrates, BS poses a diagnostic challenge because no unique clinical, laboratory or histological features have been recognised. Furthermore, some of its manifestations are common to the general population – oral ulcers – and are difficult to distinguish from other diseases – gastrointestinal involvement of BS vs CD. More than one set of classification criteria has been developed, but all have limitations when used for diagnostic purposes in an individual patient. For example, in the development of the International Study Group Criteria for Behçet Disease – one of the most widely accepted set of criteria – the control group did not include patients with CD. In this regard, it is worth noting that CD has many features that overlap with BS, namely the gastrointestinal, cutaneous, articular, ocular and cardiac manifestations (Table 1)[4].
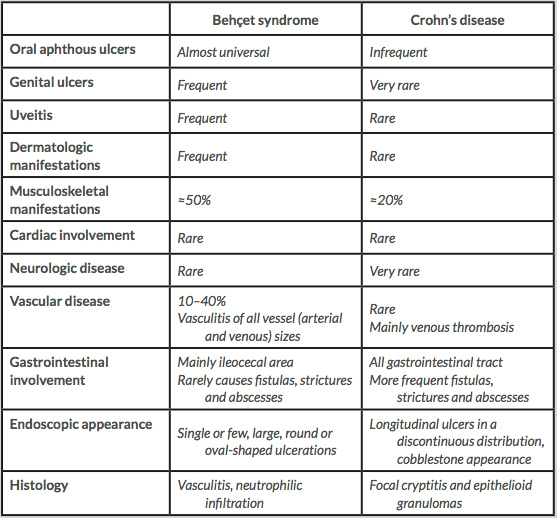
Table 1 (click to enlarge)
Table 1. Major differences between Behçet syndrome and Crohn’s disease
Our patient had a history of terminal ileitis, peripheral and axial polyarthritis, and myopericarditis, all of which can be found in BS and CD.
It may be particularly challenging to distinguish between intestinal BS and CD, but there are some useful differences between the two, namely:
(1) the finding of single or few, large, discrete, and round- or oval-shaped ulcerations in the ileocecal area suggests BS, as opposed to the longitudinal ulcers (defined as ≥4 to 5 cm) in a discontinuous distribution, cobblestone appearance, and/or small aphthous ulcerations arranged in a longitudinal fashion that suggest CD;
(2) a mucosal biopsy showing vasculitis, neutrophilic infiltration, fibrinopurulent exudates and necrotic debris indicates BS and contrasts with the classic findings of CD – discontinuous cryptic architectural abnormalities, discontinuous inflammation, focal cryptitis, and epithelioid granulomas; and
(3) a much higher incidence of fistula, stricture and abscess formation is found in CD than in BS[4].
In our patient, the endoscopic and pathologic findings were highly unspecific, which precluded an early diagnosis of BS and diverted the clinician towards the more frequent diagnosis of CD. However, the patient did well for 9 years while on sulfasalazine monotherapy – which would be highly atypical for CD – and ultimately presented with venous thrombosis and peripheral artery aneurysms, which are highly suggestive of BS. Other features that may be particularly helpful in establishing the diagnosis of BS, and are not considered in any of the classification criteria presently available, are brainstem atrophy, pulmonary arterial aneurysms, and the simultaneous finding of smooth-layered hypopyon, superficial retinal infiltrate with retinal haemorrhages and branch retinal vein occlusion with vitreous haze[2].
Treatment of BS
Treatment of BS is largely dictated by the type and severity of organ involvement. There is controversy regarding the efficacy of 5-amino-salicylates (5-ASA) in CD, but these can be used to treat gastrointestinal BS if clinical and endoscopic activity are mild[4]. Other options to treat gastrointestinal BS, besides steroids, include thalidomide, thiopurines and anti-tumour necrosis factor-α (anti-TNF), which largely overlaps with the treatment of CD, although anti-TNF therapy seems to be more efficacious in CD[4]. In our case, the initial diagnosis of CD prompted the controversial commencement of sulfasalazine. Nonetheless, this medication turned out to be effective which, in retrospect, reinforces our clinical impression that BS was the correct diagnosis from the beginning. As such, in this case, the initial misdiagnosis of CD did not have a significant clinical impact. Conversely, vascular disease in BS mandates aggressive therapy. The combination of bypass surgery and intravenous cyclophosphamide is commonly the first choice to treat BS-related peripheral artery aneurysms, which diverges from the typical therapy for CD[3,4,5]. Because recurrence at the site of anastomosis is possible, prolonged immunosuppressive therapy and frequent monitoring are necessary[3,5]. The role of anticoagulation to treat BS-related venous thrombosis is still a matter of debate, because the main driver of the thrombosis is venous wall inflammation, and this should be addressed with immunosuppressive therapy.