ABSTRACT
Introduction: Immunoglobulin A vasculitis (IgAV) is a small-vessel vasculitis with IgA-dominant immune deposits. IgAV frequently involves the skin, gastrointestinal tract, joints and kidneys. In contrast to other types of small-vessel vasculitis, IgAV is rarely complicated by intra-alveolar haemorrhage (IAH).
Methods/Results: We describe a patient with relapsing bladder cancer who presented with IAH during the course of IgAV successfully treated with corticosteroids alone.
Conclusion: This case report reminds us that IgAV can manifest with IAH. There are no robust data to support the systematic use of cyclophosphamide or plasma exchange as first-line therapy for IgAV with IAH.
LEARNING POINTS
- Intra-alveolar haemorrhage in IgA vasculitis is an uncommon but important condition.
- The treatment strategies for IgA vasculitis and intra-alveolar haemorrhage and their rare association are discussed with reference to the literature.
KEYWORDS
IgA vasculitis, Henoch-Schönlein purpura, intra-alveolar haemorrhage, cancer
CASE DESCRIPTION
An 82-year-old man was referred for an acute rash predominantly localized to the lower limbs, a 1-week history of symmetrical arthralgia of the wrists and ankles, and recent increasing dyspnoea. His past medical history included a 20-year history of tobacco use, pulmonary tuberculosis 40 years previously, and bladder cancer 25 years previously.
The patient’s vital signs were as follows: blood pressure 150/95 mmHg, temperature 37.3°C, heart rate 80/min, respiratory rate 20/min, and percutaneous oxygen saturation 94% without oxygen inhalation. Physical examination revealed a purpuric rash limited to the lower limbs, diffuse wheezing and no obvious arthritis or signs of phlebitis.
Typical biological examinations showed serum electrolytes, creatinine and proteinuria within the normal ranges; C-reactive protein was increased (89 mg/l; normal range <5), haemoglobin was decreased (9.2 g/dl; normal range >13), and platelet count was increased (50×104/μl; normal range <40).
Blood and urine cultures and other serum investigations, including anti-nuclear antibody (ANA), anti-neutrophil cytoplasmic autoantibody (ANCA), rheumatoid factor (RF), anti-cyclic citrullinated peptide antibody (anti-CCP), cryoglobulinemia, HIV and hepatitis B and C, were all negative. Complement factor CH50 was increased, while C3 and C4 were within the normal range. The plasma IgA level was increased (4.29 g/l; normal range 0.70–3.12 g/l). A skin biopsy of the rash showed leukocytoclastic vasculitis (Fig. 1A,B) and perivascular IgA deposits (Fig. 1C).
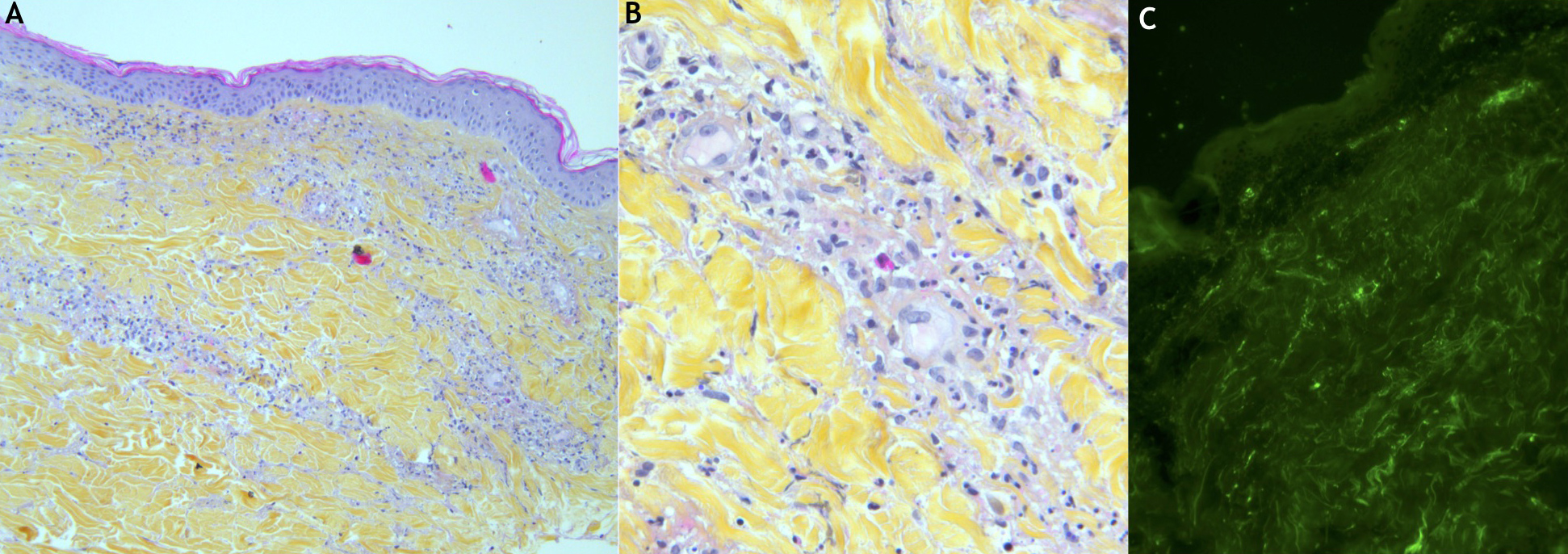
Figure 1 (click to enlarge)
Figure 1. Skin histopathology (H&E) showing leukocytoclastic vasculitis (A, B). Skin indirect immunofluorescence histopathology showing perivascular IgA deposits (C)
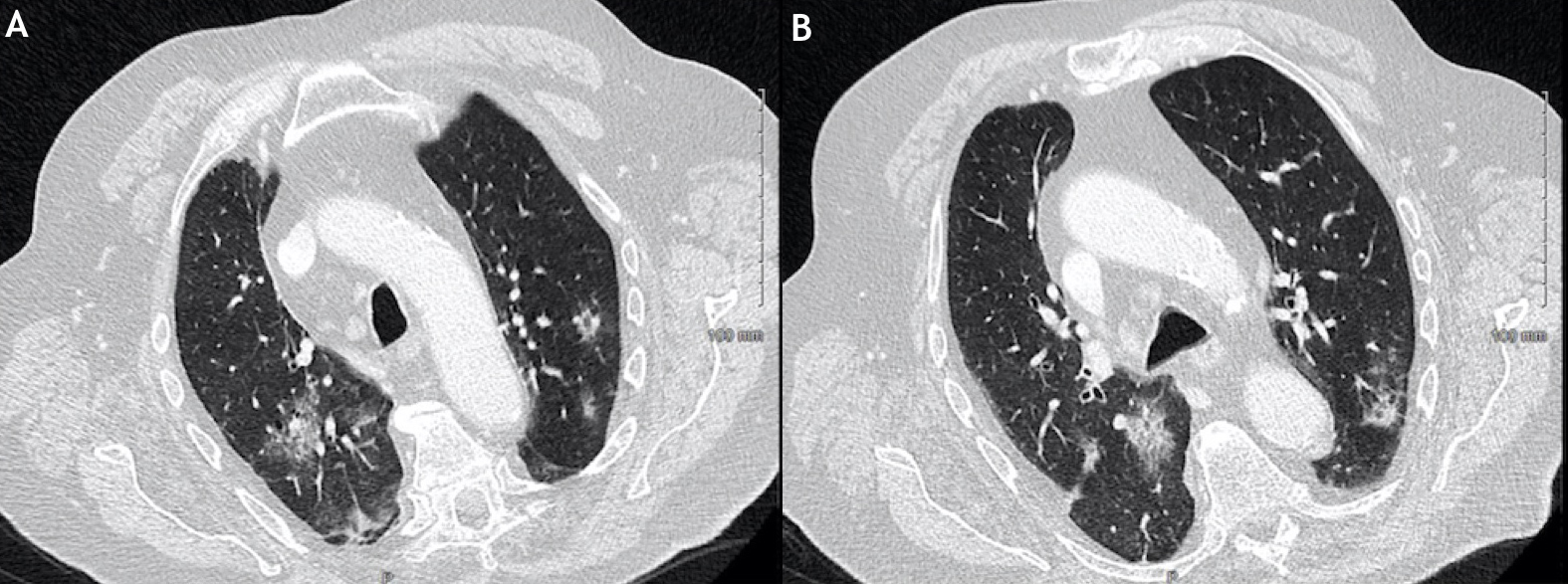
Figure 3 (click to enlarge)
Figure 2. Thoracic computed tomography (CT) scan showing focal and bilateral parenchymatous infiltrates (ground grass)
Brain natriuretic peptide was within the normal range, repeat electrocardiograms remained normal, and transthoracic echocardiograms did not reveal any cardiac dysfunction. Bronchoscopy was normal, while bronchoalveolar lavage (BAL) indicated alveolar haemorrhage with massive haemosiderin-laden macrophages and a Golde score of 258 (normal range <100) with negative direct and culture-based tests for viruses, fungi and bacteria. Based on these data, a diagnosis of IgA vasculitis with cutaneous, articular and pulmonary (intra-alveolar haemorrhage) involvement was made. A few days later, the patient had an episode of macroscopic haematuria, and cystoscopy and a CT scan revealed bladder cancer relapse. However, all clinical signs of IgAV and CT signs of IAH promptly disappeared with administration of methylprednisolone pulses (500 mg/day, 3 days) followed by tapering daily oral prednisone (1 mg/kg/day), and the patient was discharged on day 14 of admission.
During 8 months of follow-up, no IgAV relapses occurred, corticosteroids were stopped in month 3, and the management of relapsing bladder cancer was discussed.
Figure 2 (click to enlarge)
Figure 2. Diffusion MRI (A) with ADC mapping (B) sequences: in the basilar and posterior cerebral artery territories, multiple foci of diffusion restriction (hyperintensity) with a dropped bilateral ADC signal (hypointensity) are seen
DISCUSSION
In the absence of other causes such as left heart failure, infection, other autoimmune vasculitis or toxic exposure, IAH can be an uncommon manifestation of IgAV, as reported here. Indeed, in a retrospective cohort recently studied by Audemard-Verger et al.[1], IAH was only observed in one (0.5%) of 260 adult cases of biopsy-proven IgAV. The best management strategy for IAH in the context of IgAV is not known, but the successful use of corticosteroids alone is under study, as the pulmonary involvement in this case was not severe at treatment onset.
In 2013, Rajagopala et al.[2] analyzed the treatment strategies and outcomes of the 36 cases so far reported in the English literature of paediatric and adult IAH complicating biopsy-proven IgAV. Interestingly, lung biopsies were available for 36.1% (n=13/36) of the cases and showed either leukocytoclastic vasculitis with alveolar haemorrhage (69.2%, n=10/13) or alveolar haemorrhage alone without capillaritis (31.8%, n=3/13). IgA was positive in only 50% (n=3/6) of these lesions. IAH was frequently severe; 50% of the patients required mechanical ventilation, and 27.8% of the IgAV patients who experienced IAH died. Cyclophosphamide (CYC) and pulse methylprednisolone, but not plasmapheresis, for IAH was associated with better outcomes, particularly in patients who were already receiving steroids at the time of IAH.
Our patient presented with non-severe IAH and was treated with a corticosteroid regimen without CYC or plasmapheresis, and showed a complete response.
The treatment of adult IgAV remains controversial because of the absence of a correlation between initial presentation and long-term outcome and the frequent occurrence of spontaneous remission. Gastrointestinal and renal involvement in addition to IAH is a challenging and life-threatening situation in adults, but is generally benign and self-limiting in children. Most available studies have been performed in paediatric patients with IgAV or with IgA nephropathy, but results are often extrapolated to adult IgAV. CYC has been used in patients with organ- or life-threatening manifestations of ANCA vasculitis, another form of severe relapsing vasculitis. Pillebout et al. compared corticosteroids without or with CYC in adults with severe IgA vasculitis in the only available multicentre, prospective, open-label trial. Fifty-four patients with biopsy-proven IgA vasculitis with severe manifestations, including proliferative glomerulonephritis and/or severe visceral manifestations, were included[3]. At 12 months, no differences were found between the two groups regarding remission rates, renal outcomes, deaths or adverse events.
Data on plasmapheresis are scarce in IgAV. In the context of ANCA vasculitis, the PEXIVAS survey recently showed that plasma exchange does not reduce the risk of end-stage renal disease or death in patients with ANCA-associated vasculitis. Compared with a standard dose, a reduced dose of glucocorticoids did not substantially increase the risk of death or end-stage renal disease and resulted in fewer serious infections[4].
In conclusion, this case report reminds us that IgAV can manifest with IAH. Although IAH can be a life-threatening manifestation of IgAV, thus far, there are no robust data to support the systematic use of cyclophosphamide or plasma exchange in first-line therapy; prospective and controlled studies the severity of IAH are required to define the best treatment strategy in this context.