ABSTRACT
Synovitis, acne, pustulosis, hyperostosis and osteitis (SAPHO) syndrome is a rare, unrecognized and chronic inflammatory disorder characterized by distinct cutaneous and osteoarticular manifestations. Renal complications are uncommon. We describe the unusual case of a patient with severe refractory and chronic hidradenitis suppurativa (HS) that progressed to chronic renal failure due to secondary amyloidosis, presenting with mandibular pain and renal failure. The challenging diagnosis of SAPHO syndrome was made. The purpose of this case report is to emphasize the need for vigilance, timely recognition and multidisciplinary treatment. The fundamental management of AA amyloidosis and SAPHO syndrome requires an individualized approach with control of the underlying inflammatory disease.
LEARNING POINTS
- Systemic amyloidosis is a serious but rare complication of chronic inflammatory disorders such as hidradenitis suppurativa and should be periodically considered.
- SAPHO syndrome should be suspected in patients with cutaneous and osteoarticular manifestations, and is a challenging diagnosis with exclusion of neoplastic, autoimmune and infectious diseases.
- The cornerstone of management of AA amyloidosis is vigilance, timely recognition and sometimes aggressive immunosuppressive treatment.
KEYWORDS
AA amyloidosis, SAPHO syndrome, hidradenitis suppurativa
CASE DESCRIPTION
A 47-year-old man with a medical history of smoking and chronic hidradenitis suppurativa (HS) presented to the emergency department complaining of asthenia, vomiting, watery diarrhoea and a painful swelling in the left mandibular region that had started 3 weeks previously. He was self-medicated with ibuprofen 600 mg (4 times a day). He did not report fever, arthralgia or urinary symptoms.
HS had started 20 years previously with cervical, axillar and inguinal involvement. The patient had undergone plastic surgery of the right axilla and reported periods of exacerbation controlled with non-steroidal anti-inflammatory drugs (NSAIDs).
On examination, he was debilitated, normotensive and apyretic. Cardiac, pulmonary and abdominal evaluations did not show any alterations. Examination of the skin revealed confluent inflammatory nodules with purulent drainage (Hurley 3/3) in the gluteal area (Fig. 1).

Figure 1 (click to enlarge)
Figure 1. Skin examination revealing confluent inflammatory nodules with purulent spontaneous drainage (Hurley 3/3) in the gluteal and axillar regions
Arterial blood gas revealed severe metabolic acidaemia (pH 7.22, HCO3 8.3). Laboratory findings reported normocytic anaemia (Hgb 6.7 g/dl; normal 13.5–18.0 g/dl), leucocytosis (13.87 K/µl; normal 4–10 K/µl), uraemia (322 mg/dl; normal 6–23 mg/dl) and elevated creatinine (20.55 mg/dl; normal 0.50–1.20 mg/dl), with low-normal albumin (3.5 g/dl; normal 3.5–5.2 g/dl) and hyperkalaemia (5.1 mmol/l; normal 3.4–5.0 mmol/l). The urinary study showed a protein to creatinine ratio of >4.00 mg/mg. An abdominal and renal ultrasound revealed splenomegaly (18 cm bipolar axis), kidneys with preserved dimensions and increased cortico-medullar differentiation and absence of hydronephrosis. The patient started urgent renal replacement therapy with haemodialysis and was admitted to the internal medicine department.
The patient maintained haemodialysis with no recovery of renal function. A 24-hour urine collection documented proteinuria of 18.7 g/day. A renal biopsy with immunostaining study was performed, revealing deposition of amyloid A in all 34 glomeruli and vascular walls with apple-green birefringence upon examination of Congo red-stained tissue under polarized light microscopy (Fig. 2).
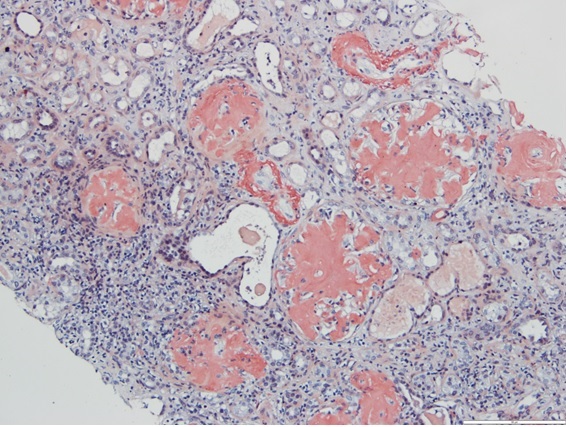
Figure 2 (click to enlarge)
Figure 2. Renal biopsy with Congo-Red stain revealing orange-red deposits in all 34 glomeruli and vascular walls
AA was confirmed by immunohistochemical amyloid protein identification with DNA analysis. Infectious, autoimmune (including anti-nuclear antibody, rheumatoid factor, anti-Ds-DNA, anti-ANCA and anti-citrullinated peptide antibody) and monoclonal gammopathy (including serum protein electrophoresis, urine protein electrophoresis, immunofixation and serum free light chain assay) studies were negative. Inflammatory studies revealed an elevated erythrocyte sedimentation rate (ESR) of 119 mm/h, C-reactive protein (CRP) of 152.2 mg/l and serum amyloid protein (SAP) of 11.9 ng/l (normal 0.0–6.4 ng/l). Coagulation and cardiac studies (including NT-proBNP and troponin levels) were normal.
As the patient maintained watery diarrhoea without an infectious cause identified and to evaluate for another manifestation of amyloidosis, a colonoscopy with biopsy was performed and showed amyloid deposition (Fig. 3) without signs of inflammatory bowel disease. A transthoracic echocardiogram and ECG did not reveal any alterations.
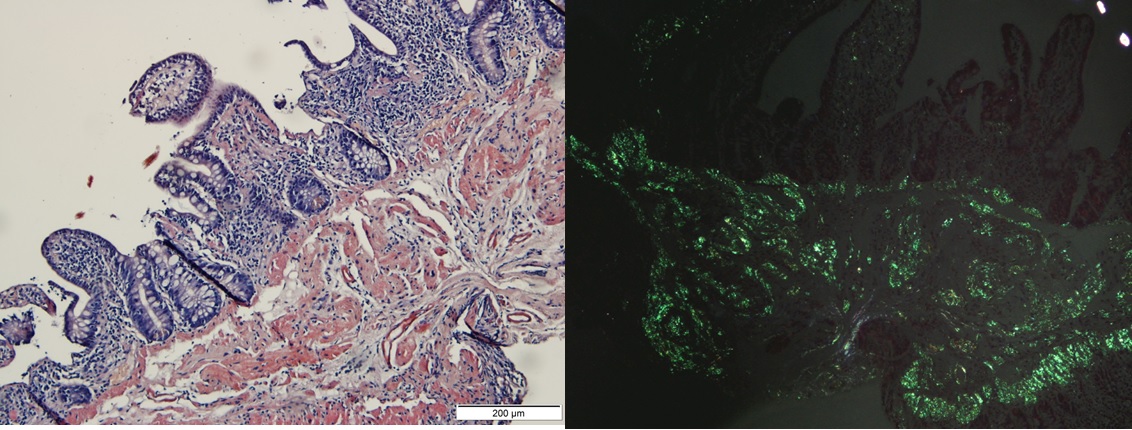
Figure 3 (click to enlarge)
Figure 3. Left panel: ileal biopsy revealing the presence of deposits of an amorphous substance on the ileal mucosa, predominantly in the submucosa and vessel wall, highlighted by Congo red staining (200×). Right panel: ileal biopsy showing apple-green birefringence on observation with polarized light
As HS is associated with inflammatory rheumatic diseases and an increased risk of malignancy, a mandibular and thoracoabdominal computed tomography (CT) scan was performed and revealed sclerosis with cortical periosteal reaction in the left mandibular branch, subchondral sclerosis in the right sacroiliac joint and hyperostosis at the proximal extremities of the clavicles and in some sterno-costal joints (Fig. 4).
A bone marrow biopsy with immunocytochemical study also documented deposits of amyloid A but no evidence of lymphoproliferative or plasmocytic disease.
Due to the association of HS with radiological multifocal (mandibular, sternoclavicular, sacroiliac, vertebral) osteoarticular findings, SAPHO syndrome was suspected. Therefore, 99mTc bone scintigraphy was performed and showed multifocal (sternocostoclavicular, sacroiliac joints, left mandibular branch) hypercaptation (Fig. 5). A mandibular biopsy documented no evidence of neoplastic disease.

Figure 4 (click to enlarge)
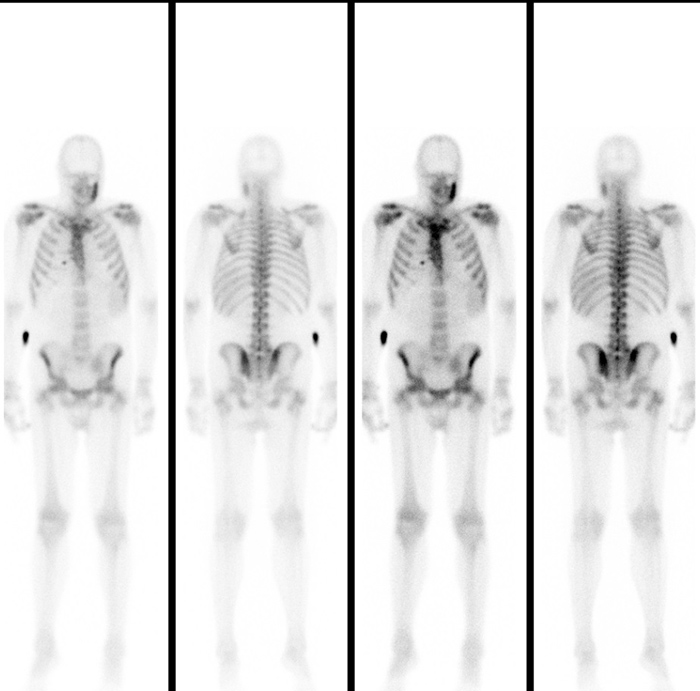
Figure 5 (click to enlarge)
Figure 4. Thoracic x-ray showing hyperostosis at the proximal extremities of the clavicles and in some sterno-costal joints
Figure 5. Full-body bone scintigraphy revealing multifocal (sternocostoclavicular, sacroiliac joints, left mandibular branch) hypercaptation of the radiopharmaceutical agent
The patient maintained confluent inflammatory nodules with purulent drainage (Hurley 3/3) in the gluteal area, and was medicated with doxycycline 100 mg. As the skin lesions worsened after 2 months, biological treatment with adalimumab was considered.
DISCUSSION
This case report is remarkable due to the rarity, sequential and systemic manifestations, and severe evolution of the condition, and the diagnostic approach undertaken. It describes a patient with a 20-year history of HS controlled with NSAIDs, who presented with mandibular pain and terminal renal failure. The diagnostic investigation revealed systemic amyloidosis (renal, intestinal and bone marrow). SAPHO syndrome was also diagnosed based on the chronic HS and multifocal signs of osteitis and hyperostosis (especially with uncommon mandibular involvement) after anatomo-pathological findings and exclusion of infectious, malignant or other aetiological disorders[1].
SAPHO syndrome is a rare, frequently misdiagnosed, relapsing and inflammatory disorder of uncertain aetiology characterized by distinct cutaneous and osteoarticular manifestations. SAPHO syndrome should be suspected in patients with bone and joint symptoms consistent with inflammatory osteitis, especially of the anterior chest wall (involved in 65–90% of cases) and sacroiliac joints, associated with neutrophilic dermatosis or acneiform eruption. The mandibular involvement is rare[2].
HS is a recurrent disease associated with inflammatory, rheumatological and neoplastic diseases and therefore requires a multidisciplinary approach[3].
This case is also unusual as the patient presented with advanced renal failure associated both with AA amyloidosis secondary to the chronic hidradenitis, and also with chronic anti-inflammatory treatment with NSAIDs.
Systemic AA amyloidosis is a serious but rare complication of chronic inflammatory disorders with end-stage kidney failure a predominant cause of death. Renal involvement should be periodically screened for with a search for proteinuria.
The cornerstone of management of SAPHO syndrome and AA amyloidosis is to control the underlying inflammatory disease, which requires individualized treatment guided by symptoms and based on the few cases reported. Patients with an inadequate response to a unified approach to therapy (retinoids or antibiotics) are controlled with methotrexate or a TNF inhibitor[4]. However, patients with refractory HS showed a specific response to adalimumab, which is being considered for patient treatment[5]. We emphasize the importance of limiting the adverse events associated with anti-inflammatory drugs and the emergence of new therapies to promote the clearance of amyloid deposits of different types.