ABSTRACT
Langerhans cell histiocytosis (LCH) is a multisystemic disorder that results from the clonal proliferation of immunophenotypically and functionally immature Langerhans cells (LC). The detection of the V600E mutation in the BRAF oncogene in LCH biopsy specimens supports previous evidence that LCH is a neoplastic disorder. This mutation is present in other cutaneous lesions including malignant melanoma and benign nevi. Single case reports of a correlation between LCH and the appearance of eruptive nevi limited to the inguinal folds after chemotherapy have previously been described in the literature. This suggested that LCH could be an additional cause of eruptive melanocytic nevi, with a specific distribution mimicking that of LCH cutaneous lesions. We present the case of a 6-year-old boy, previously treated with chemotherapy for Langerhans cell histiocytosis, with disseminated junctional nevi. Although this co-occurrence may be coincidental, the skin involvement is distinct from other previously reported clinical cases. It would be interesting to evaluate whether the BRAF mutation described in LCH cells might in fact support a genetic background for the development of nevi in these patients.
LEARNING POINTS
- Langerhans cell histiocytosis (LCH) is a clonal neoplastic proliferation of immature Langerhans cells, with the V600E mutation in the BRAF oncogene present in approximately 60% of cases.
- The V600E mutation in the BRAF oncogene is also documented in other cutaneous lesions, namely malignant melanoma and benign nevi.
- There are case reports of a correlation between LCH and the appearance of eruptive nevi after chemotherapy, but it is not known whether the BRAF mutation described in LCH cells supports a genetic background for the development of nevi in these patients.
KEYWORDS
Langerhans cell histiocytosis, eruptive nevi, chemotherapy, neoplastic disorder
INTRODUCTION
Langerhans cell histiocytosis (LCH) is a clonal neoplastic proliferation of immunophenotypically and functionally immature, morphologically rounded Langerhans cells along with eosinophils, macrophages, lymphocytes, and occasionally, multinucleated giant cells[1]. It is a multisystemic disease that that ranges in severity from a fatal leukaemia-like disorder to an isolated lytic bone lesion[2]. Although controversy persists whether the proliferation of LC results from a malignant transformation or is the result of an immunological stimulus[2–4], the discovery that approximately 60% of LCH biopsy specimens harbour the V600E mutation in the BRAF oncogene reinforced the evidence that LCH is a clonal neoplastic disorder, challenging the concept of it being a reactive disorder[5,6]. The presence of this mutation is also well established in other cutaneous lesions including malignant melanoma and benign nevi[6].
Patients may present with single organ disease (single-system LCH), which may involve a single site (unifocal) or multiple sites (multifocal). Treatment decisions are based upon whether high-risk (liver, spleen and bone marrow) or low-risk organs (skin, bone, lung, lymph nodes, gastrointestinal tract, pituitary gland and the central nervous system) are involved and whether LCH presents as single-system or multisystem disease. Depending on the site and extent of disease, treatment of LCH may include surgery, chemotherapy, radiation therapy or topical therapy[7]. Case reports have described activity of BRAF inhibitors against LCH in paediatric patients, but there are insufficient data to validate the role of these agents in the treatment of children with LCH[7,8]<7sup>.
CASE REPORT
A 6-year-old boy with a history of LCH with skin and bone involvement diagnosed at the age of 6 months was referred to our outpatient clinic due to the appearance of multiple nevi. He was initially treated with vinblastine and prednisolone, according to the LHC-III protocol (group 2), but due to disease recurrence with pituitary gland involvement, cytarabine (six cycles) and indomethacin as maintenance therapy were administered for 2 years.
While on maintenance therapy, after 15 months of indomethacin, the patient developed pigmented lesions over his whole body, sparing only the head and acral regions. Physical examination (Fig. 1) revealed small, 1–4 mm in diameter, asymptomatic brown macules with a fine peripheral network and small globules on dermoscopy (Fig. 2). Histological examination (Fig. 3) confirmed the diagnosis of junctional nevus. The patient is currently under follow-up in our outpatient clinic.
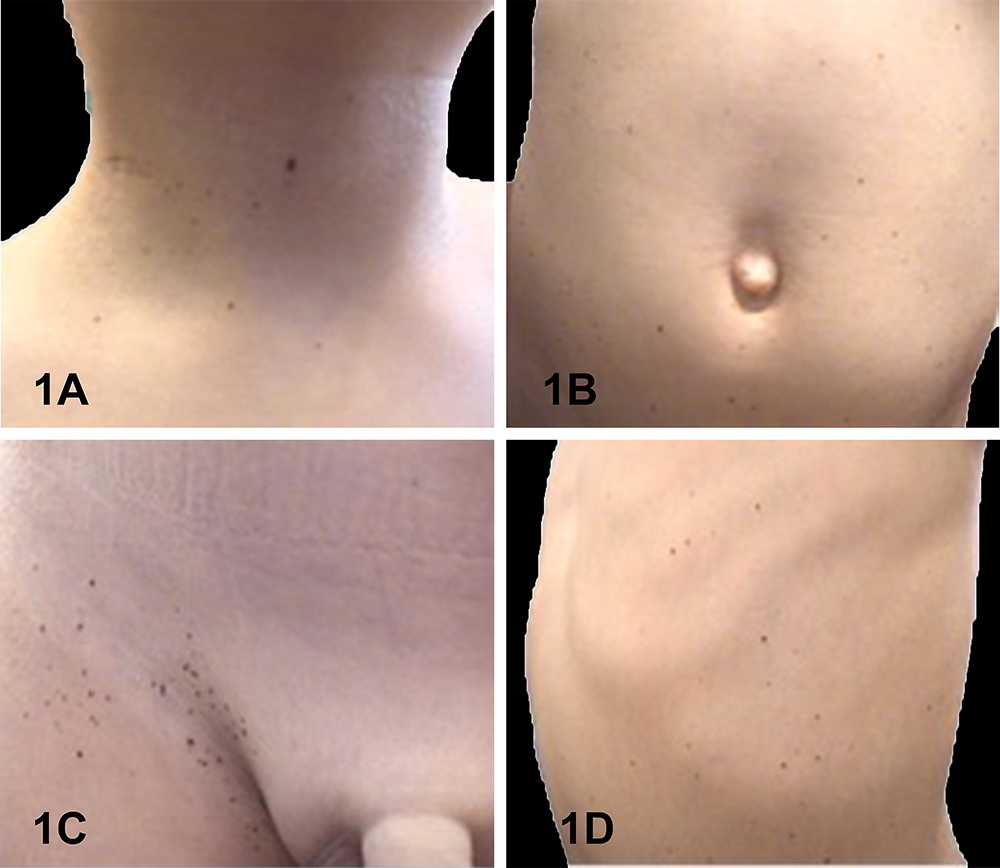
Figure 1 (click to enlarge)

Figure 2 (click to enlarge)
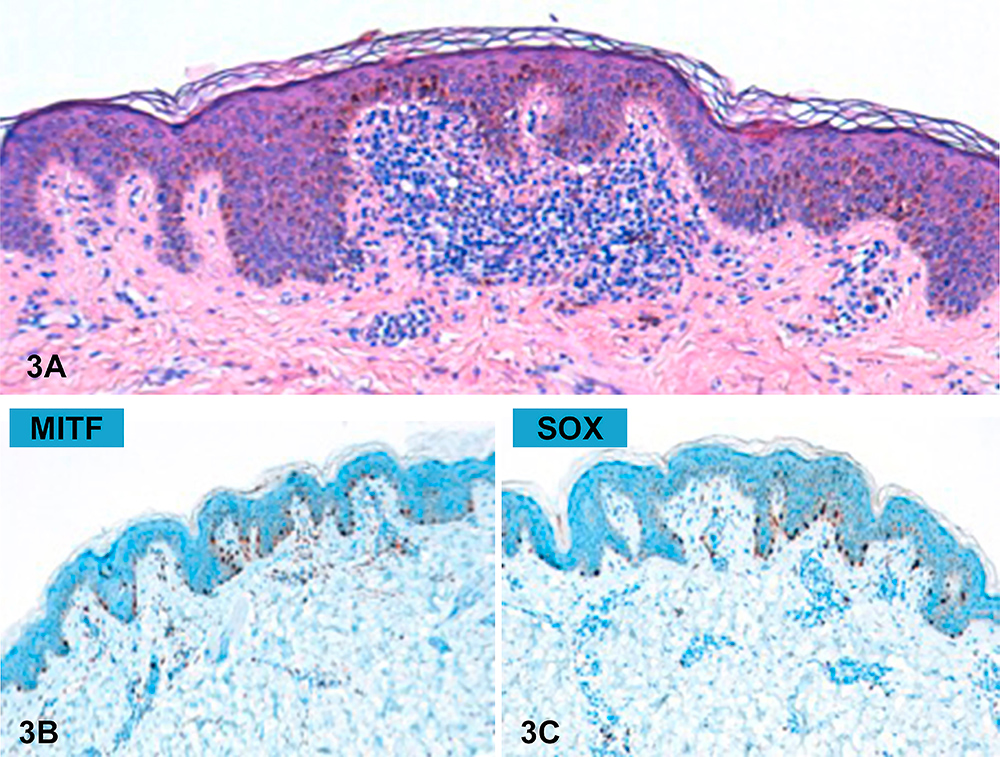
Figure 3 (click to enlarge)
Figure 1. Clinical images of multiple brown macules over different anatomical areas: (1A) neck; (1B) abdominal wall; (1C) right inguinal fossa; (1D) left flank
Figure 2. Dermatoscopic features: (2A) fine peripheral network; (2B) small globules (arrow head)
Figure 3. (3A) Histological features (H&E); (3B) MITF expression; (3C) SOX expression
DISCUSSION
Berk et al.[9] and Surinach et al.[10] have previously reported a correlation between LCH and eruptive nevi. However, in these cases the lesions were limited to the inguinal folds. The latter authors suggested that LCH could be an additional cause of eruptive melanocytic nevi, with a specific distribution mimicking that of LCH cutaneous lesions, and that chemotherapy might act as a precipitating factor[10]. This suggestion is supported by the fact that an increased number of regulatory T cells are seen in LCH lesions. As such, this could result in the appearance of eruptive nevi by promoting immune tolerance[11]. It may be also possible that proinflammatory cytokines secreted in LCH lesions, such as interleukin (IL)-1, granulocyte-macrophage colony-stimulating factor, interferon-γ and IL-4, may promote nevi development[12]. The up-regulation of melanocortin-1 receptor (MC1R) mRNA expression by IL-1 may also play an important role in nevi development since melanocyte-stimulating hormone α, the MC1R ligand, promotes nevogenesis[13,14]. This clinical case is somewhat distinctive, since the lesions are disseminated, arguing against the above-mentioned hypotheses. Although this co-occurrence may be coincidental, it is interesting to consider that since the BRAF mutation has been described in LHC cells, this might in fact support a genetic background for the development of nevi in these patients. Even though it is well recognized that there is a need for additional mutations to render the cell malignant, as V600E-mutated BRAF is also present in benign nevi, it would be interesting to evaluate the potential development of melanoma in these cases with close clinical follow-up.