ABSTRACT
Background: Klinefelter syndrome (KS) is the most common sex-chromosomal disorder in males. Frequently under-recognized, it occurs in 1 in 500–600 male births. It is caused by the inheritance of at least one additional X chromosome from either parent. Patients often have uncommon or atypical malignancies.
Patient: We describe the case of a 35-year-old man with 47XXY KS and previous cryptorchidism, presenting with a painful testicular mass. Histology confirmed Leydig cell hyperplasia.
Discussion: Cryptorchidism is an established risk factor for testicular tumours and occurs six times more commonly in KS than in the general population. Despite this, large epidemiological studies have shown a reduced burden of testicular cancer in these patients. The presentation of a hypoechoic lesion on ultrasound will prompt consideration of testicular tumours, however orchalgia represents an atypical presentation. In patients with KS, Leydig cell hyperplasia is a much more common entity and should be considered early in the differential diagnosis.
LEARNING POINTS
- The differential diagnosis of a testicular mass in Klinefelter syndrome includes malignancy and nodular Leydig cell hyperplasia.
- Diagnosis can be challenging, both radiologically and histologically.
- Orchalgia is atypical in Leydig cell hyperplasia.
KEYWORDS
Leydig cell hyperplasia, Klinefelter syndrome
CASE DESCRIPTION
A 35-year-old man with a known history of Klinefelter syndrome (KS) presented to the endocrine service with a 2-week history of left-sided testicular pain.
He was diagnosed with KS at the age of 15 years when he attended his general practitioner with a 2-year history of gynaecomastia and small bilateral testes were noted. He had a background history of a left-sided undescended testis and orchidopexy aged 3 years. Physical examination showed a height of 183 cm, weight 54.3 kg (BMI 16.2 kg/m2) and a testicular volume of 2 ml with marked gynaecomastia. Serum testing results for follicle stimulating hormone (FSH) and luteinizing hormone (LH) were elevated at 61.0 IU/l and 41.0 IU/l, respectively. The prolactin level was 330 µg/l (80–300) and chromosomal analysis confirmed a 47XXY karyotype. He was commenced on testosterone intramuscularly and subsequently had bilateral subcutaneous mastectomy with a good cosmetic result.
At the time of this presentation he had no other medical conditions. He was in a stable relationship and a recent sperm analysis confirmed azoospermia (sperm count zero). This patient had previously been stable on intramuscular testosterone undecanoate, but switched to transdermal testosterone after developing pulmonary microembolism.
METHODS AND PROCEDURES
Examination revealed small testes (volume <5 ml) bilaterally. The left testis was swollen, erythematous and tender to palpation. Testicular ultrasound was performed and confirmed bilateral atrophic testes and a 5 mm hypoechoic lesion in the upper pole of the left side (Fig. 1, arrow), highly suspicious for tumour.
Serum samples for the testicular tumour markers alpha-fetoprotein (range 0–9 µg/l) and human chorionic gonadotropin (0.0–5.0 IU/l) were <2 µg/l and <1.2 IU/l, respectively. A pre-operative CT of the thorax, abdomen and pelvis was unremarkable.
The conclusion of the urology multidisciplinary meeting was that the appearance was very suspicious of tumour and the patient underwent a left radical inguinal orchidectomy.
Histology revealed a field change of nodular Leydig cell hyperplasia (LCH), 20 mm in maximum dimension, on a background of testicular atrophy, consistent with the history of cryptorchidism. A large nodular focus of Leydig cells, 4 mm in diameter, with associated atrophic tubules, was present and appeared to correlate with the hypoechoic lesion identified on ultrasound examination. The epididymis and spermatic cord were both normal and there was no evidence of malignancy (Fig. 2).
The patient had an uncomplicated recovery and remains clinically well 18 months post-operatively. He was advised to undertake self-examination of the right testis.
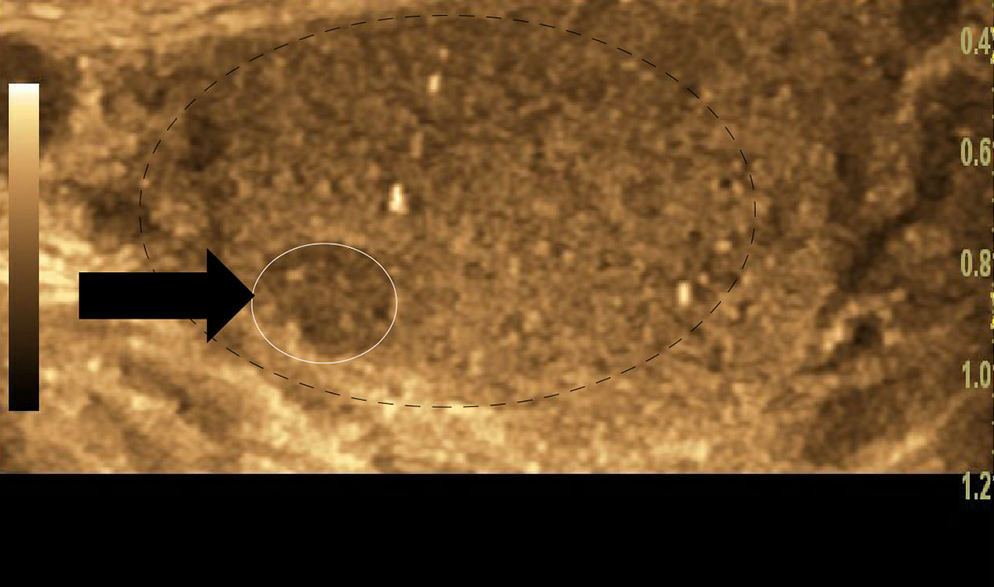
Figure 1 (click to enlarge)

Figure 2 (click to enlarge)
Figure 1. A hypoechoic lesion measuring 5 mm in maximum diameter (indicated by a black arrow, and outlined with a dashed white line) confined to the upper pole of the left-sided testis, which is otherwise atrophic (outlined with a broken black line)
Figure 2. Hyalinised seminiferous tubule enveloped in a nodule of Leydig cells, and background testicular atrophy (H&E, 40×)
DISCUSSION
Klinefelter syndrome (KS) is a sex chromosomal disorder typically presenting with gynaecomastia, aspermatogenesis and elevated gonatrophins.
The most pertinent differential diagnoses of a testicular mass in KS include LCH or tumour and malignancy. Establishing the diagnosis is often challenging both radiologically and histopathologically; either condition can present with hypo-echogenicity on ultrasound and there is insufficient experience with hyperplasia visualised on MRI to differentiate the two entities[1]. The over-lapping histopathological features include proliferation of acidophilic cells[2]. In this case, the multifocality of the Leydig cell proliferation, with no individual focus being greater than 5 mm, along with the interstitial growth and entrapped seminiferous tubules, favoured a diagnosis of nodular Leydig cell hyperplasia rather than a Leydig cell tumour, and there was no evidence of malignancy.
Germ cell depletion, seminiferous tubule hyalinisation and fibrosis have been demonstrated histologically in KS. From adolescence onwards, the testicular changes progress rapidly and severe LCH is seen[3]. These areas of hyperplasia may be identifiable as micro-calcifications, nodules or cysts on ultrasound.
Like LCH, testicular cancer was an important consideration given the patient’s presentation and cryptoorchidism. Cryptoorchidism occurs in up to 31% of KS due to a reduced testosterone level during embryonic life. Additionally, 10% of all cases of germ cell tumours will occur in patients with cryptoorchidism or a previous history of such. Considering this, it would appear logical that KS patients would have a higher risk of testicular tumours. However, two large epidemiological studies (including over 4,000 karyotypically confirmed patients with KS from the UK and Denmark have conclusively shown otherwise[4].
This case is also novel as the patient presented with orchalgia. In much of the previously described literature, microcalcifications or hyperplasia has been found on routine investigation of radiology performed for research purposes. To our knowledge only one other case of LCH presenting with orchalgia has been reported[5].
CONCLUSION
In summary, LCH remains an important differential diagnosis when a patient with KS and a testicular mass is being investigated. While it is difficult to differentiate conditions pre-operatively, more conservative surgery may be considered if LCH is confirmed and as such it may alter patient outcome.