ABSTRACT
Objectives: Thrombocytopenia and splenomegaly are common features in several haematological disorders. Gaucher disease (GD) is a rare lysosomal storage disorder frequently characterized by thrombocytopenia and splenomegaly, which represents a clinical challenge for haematologists and internists.
Case: We describe the case of a 37-year-old patient with a diagnosis of spherocytosis since childhood, who developed hepatic failure and presented striking features of GD including hepatosplenomegaly, bone fractures and post-partum bleeding. We reconsidered the diagnosis of spherocytosis and investigated Gaucher disease.
Conclusion: GD should be considered in the differential diagnosis of thrombocytopenia and splenomegaly.
LEARNING POINTS
- Although rare, Gaucher disease should be taken into consideration in differential diagnosis of splenomegaly and/or thrombocytopenia.
- Accumulation of glucocerebroside can occur in several organs (bone marrow, spleen, liver, skeleton, and rarely lungs and central nervous system), leading to multiorgan damage and clinical complications that may mislead the diagnosis.
- Early diagnosis is extremely important because ERT is effective in preventing or reversing many manifestations, including splenomegaly, bone marrow infiltration,cytopenia and osteopenia.
KEYWORDS
Spherocytosis, Gaucher disease, splenomegaly, lysosomal disorder, thrombocytopenia
CASE REPORT
A 37-year-old Caucasian woman presented to the emergency department with gradually increasing abdominal girth, weight gain (3 kg in 6 days) and swelling in the ankles. She complained fatigue and exertional dyspnoea. She denied any alcohol use or spontaneous bruising. No history of liver diseases or hepatitis was reported. She referred a diagnosis of spherocytosis for persistent anaemia (mean haemoglobin value 10 g/dl) with occasional blood transfusions, thrombocytopenia (100,000/mm3) and hepatosplenomegaly at the age of 3 years. She reported a delayed growth during childhood and a severe fracture of the left humerus, associated with humeral head necrosis, playing basketball when she was a teenager. No further investigations were performed at that time. The last MR study was performed in 2009, demonstrating left humeral head osteonecrosis associated with severe arthrosis and fibrocystic dysplasia of the proximal third of the diaphysis of the humerus. She had two pregnancies, at the age of 30 and 33 years. Post-partum bleeding occurred after the first delivery, with a need for blood transfusion, in the absence of blood coagulation abnormalities and history of prothrombotic diseases.
At admission, physical examination revealed pallor, hepatosplenomegaly (liver at 6 cm and spleen at 10 cm from the lower costal margin) and oedema in both legs. She was admitted to the internal medicine department for further investigation.
Blood examination showed leukopenia (1,730/mm3) with reduced neutrophil count (1,003/mm3), thrombocytopenia (92,000/mm3) and normochromic normocytic anaemia (red blood cells [RBC] 2,450/mm3, Hb 7.8 g/dl, mean corpuscular volume [MCV] 90.1 fl, mean corpuscular haemoglobin [MCH] 27.4 pg, red cell distribution width [RDW] 15.9%) with mild increased ferritin value (352 ng/ml) and normal transferrin saturation and serum iron (21% and 34 mcg/dl, respectively). Haemolytic indices, folates and vitamin B12 were all normal. Because of the previous diagnosis of spherocytosis, blood smear and osmotic globular resistance tests were performed, and the results were normal, excluding the diagnosis of spherocytosis. Renal and liver function were within the normal limits, except for reduced pseudocholinesterase (2,760 U/l, normal range 5,300–12,900) and hypergammaglobulinaemia at the protein electrophoresis with immunofixation positive for immunoglobulin G lambda (0.60 g/dl), increased beta2-microglobulin (4.48 mcg/ml, normal range 0.6–2.6) and negative urine immunofixation. Autoimmune, virological and neoplastic diseases were excluded. Thrombophilic screening was repeated, and results were normal. JAK2-V617F mutation was absent. Testing for HFE mutations did not detect C282Y or H63D, excluding genetic haemochromatosis.
Abdominal ultrasound confirmed the splenomegaly (diameter 18 cm). The CT scan also documented a chronic hepatosteatosis with portal hypertension (diameter of the portal vein 18 mm). Hepatic stiffness assessed by transient elastometry (FibroScan®) was 7.4 KPa (normal value <5 KPa), consistent with Stage 1 fibrosis. Oesophagogastroduodenoscopy excluded oesophageal varices. Results of a chest X-ray and gynaecological examination were normal. The swelling of the ankles was completely solved with diuretics; the patient was transfused with 2 units of blood.
As the haematological data ruled out a diagnosis of spherocytosis, a liver biopsy was performed. The specimen revealed the presence of fibrosis and significant portal and lobular infiltration by aggregated polygonal cells CD68+ with cytoplasm including fibrillar material PAS+, without iron overload. These findings were suggestive of lysosomal storage disorder, including Gaucher disease (GD), which was confirmed by increased chitotriosidase activity (14,290 nmol/h/ml, normal range <100) and undetectable beta-glucosidase activity (28 nmol/h/mg, normal range 200–500). The beta-glucosidase gene sequence showed a compound heterozygosity for N370S (c.1226A>G) and W184R (c.667T>C) mutations.
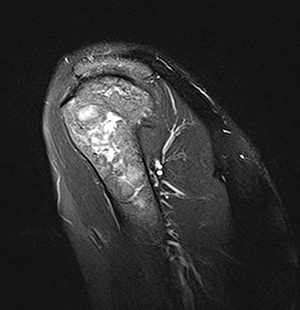
Figure 1 (click to enlarge)
Fig. 1 - Coronal T2-weighted sequence of the proximal left humerus showing severe bone alteration of the humeral head with decreased signal intensity and focal hyperintensity diagnostic of bone infarcts.
Instrumental and clinical evaluation of GD was performed. MRI of the lumbar spine, bilateral hips and left humerus showed diffuse marrow changes with bone infarcts in the left humerus consistent with GD (Fig. 1).
Endocrinological evaluation, including blood and urine tests, was normal; a DEXA scan showed a mild osteopenia (spine and femoral neck Z-scores, respectively, of –1.5 and –1.3). Lipid assessment demonstrated reduced serum cholesterol and high-density lipoprotein concentration, as commonly observed in GD. Chest X-ray and respiratory tests were normal. Electrocardiogram revealed normal sinus rhythm with frequent extrasystoles, not confirmed by 24-h ECG. Cardiac MR with gadolinium contrast excluded iron overload (T2* 28.93 ms, normal value >20 ms) and the presence of glycosphingolipids in the myocardial tissue. Pulmonary hypertension was excluded by cardiac MR and Doppler echocardiogram. Neurological evaluation excluded abnormalities, including peripheral neuronopathy, tremor and other extrapyramidal movement abnormalities and cognitive dysfunction.
The patient started enzyme replacement therapy (ERT) in July 2013 with Cerezyme® 60 U/kg every 2 weeks, and after one year of ERT the laboratory parameters had improved (Table 1).
| Variable | Reference range | At diagnosis | After one year of ERT |
|---|---|---|---|
| Leukocytes (/mm3) | 4,800–10,800 | 1,730 | 3,000 |
| Neutrophils (%) | 40-75 | 58 | 67 |
| Haemoglobin (g/dl) | 12-16 | 7.8 | 12.0 |
| MCV (fl) | 78-99 | 90.1 | 83.3 |
| Platelets (/mm3) | 130,000-400,000 | 92,000 | 146,000 |
| Serum iron (mcg/dl) | 37-145 | 34 | 62 |
| Transferrin saturation (%) | <45 | 21 | 21 |
| Serum ferritin (ng/ml) | 15-150 | 352 | 288 |
| Pseudocholinesterase (U/l) | 5,300-12,900 | 2,764 | 3,800 |
| Immunoglobulin IgG lambda (g/dl) | ND | 0.60 | 0.40 |
| Beta2-microglobulin (mcg/ml) | 0.8-2.2 | 3.4 | 1.59 |
| Chitotriosidase (nmol/h/ml) | <100 | 14,290 | 3,254 |
Table 1 - Laboratory data. ND, not detectable
Actually, the patient is well and she is still receiving ERT. No side effects have occurred, and her compliance to the therapy is satisfactory.
DISCUSSION
Anaemia and splenomegaly are common manifestations of several haematologic diseases, including haemolytic anaemias, thalassaemia and some lysosomal storage disorders such as GD. GD is the most common autosomal recessive lysosomal storage disorder, which is caused by a deficiency of the enzyme glucocerebrosidase responsible for the accumulation of glucosylceramide (glucocerebroside) in the reticuloendothelial cells of the liver, spleen and bone marrow[1]. More than 350 mutations in the glucocerebrosidase gene (GBA) have been reported, with frequencies that vary by ethnicity and disease type, leading to heterogeneous presentations and manifestations with often uncommon features. GD is a complex and systemic disease representing a real challenge for internists and haematologists[2]. Splenomegaly and/or thrombocytopenia are the most common presenting features, but other clinical manifestations include hepatomegaly, bone pain with skeletal complications, bleeding diathesis, pulmonary hypertension and neurological manifestations[3]. ERT is available and proved to be safe and effective in reversing or preventing many manifestations[4].
Starting from a simple diagnostic algorithm proposed by Mistry et al.[5], it is possible to correctly diagnose GD in the presence of splenomegaly and/or thrombocytopenia. This approach is extremely useful to avoid misdiagnosis/underdiagnosis and diagnostic delays, in order to start ERT, which can prevent severe organ damage. In our case, the correct diagnosis was made at the age of 37 years. Signs and symptoms had been present since the age of 3 years, and the patient had been incorrectly diagnosed with spherocytosis.