ABSTRACT
We report the case of a 28-year-old man who presented with recurring episodes of high fever, pleural and pericardial effusions and bilateral hydrocele. He was diagnosed with familial Mediterranean fever (FMF) and responded well to colchicine therapy. Genetic testing showed variants of the MEFV gene (p.Pro369Ser and p.Glu148Gln) previously independently described as having a more benign course of the disease. Their association is very rarely reported. Our patient and our review of the literature suggest that these genetic variants are associated with indolent courses but might also trigger the classic symptoms seen in severe FMF, probably in a compound heterozygous fashion. The combination of these variants should be taken into consideration in the diagnosis and management of patients.
LEARNING POINTS
- Familial Mediterranean fever is characterized by recurring episodes of fever with serositis, arthritis or abdominal pain.
- HMany mutations of the MEFV gene are responsible for the syndrome, but some variants are still of uncertain clinical significance.
- The p.Pro369Ser and p.Glu148Gln variants are rarely described together and can be pathogenic.
KEYWORDS
Familial Mediterranean fever, MEFV gene, p.Pro369Ser/p.Glu148Gln
CASE DESCRIPTION
We report the case of a 28-year-old man who presented with recurring episodes of unexplained fever (40°C). He complained of continuous pain in the right upper abdominal quadrant, not related to eating or drinking. He denied chest pain or cough and did not have vomiting or nausea. His intestinal transit was regular, and as were his stools and urine colour. He did not have any urinary symptoms but had noticed painful testicular oedema which resolved after he applied ice to it. Physical examination showed mild tenderness in the right upper quadrant without jaundice.
The patient was originally from Tunisia and had just returned from a recent visit there, where he had experienced an episode of fever and had been treated with levofloxacin for prostatitis. He was an active smoker, was not taking any other medication and was in good health before his trip.
He had been treated a year earlier in another city for pleural and pericardial effusions, thought to be of viral origin. Blood tests performed at the time showed a high C-reactive protein (CRP, 258 mg/l) with normal white blood cells at 7,110/mm³ with a slight neutrophil increase of 6,260/mm³ and lymphocytes down to 580/mm³. Haemoglobin was 12 g/dl and there was mild thrombocytopenia: platelets were 92,000/mm³. Renal function and hepatic tests were within the normal range. Testing for CMV, EBV, hepatitis A, B and C, and HIV came back negative. Thoraco-abdominal computed tomography (CT) revealed small bilateral pleural as well as pericardial effusions up to 12 mm in size. Hepatomegaly and splenomegaly were seen on imaging along with ascites. His echocardiography results were normal except for mild pulmonary hypertension. Scrotum ultrasound showed bilateral hydrocele without signs of epididymitis. Blood cultures and pleural effusion culture came back negative. His CRP decreased under colchicine, without any antibiotic treatment, as did all effusions. He had stopped taking colchicine during his journey in Tunisia because he ran out of medicine.
Given his country of origin and the recurring fever and symptoms without any cardiac or infectious causes found, familial Mediterranean fever (FMF) was suspected and the patient was put back on colchicine which ultimately resolved all his symptoms.
The patient presented 2 months later to our emergency department with another episode of fever, headaches and testicular oedema. He admitted stopping his colchicine treatment again.
As all symptoms including fever returned each time the patient stopped his colchicine treatment, and as he mentioned his sister had a similar medical history with remission during pregnancy, we decided to pursue genetic testing to confirm the suspicion of FMF. Unfortunately, our patient’s sibling refused to be tested for the mutations. However, genetic testing of our patient did reveal two variants of the MEFV gene: p.Pro369Ser and p.Glu148Gln. These two variants are rarely seen in the same person, and patients with these genetic variants presenting with severe FMF symptoms are seldom described in the literature.
DISCUSSION
FMF is a periodic fever syndrome defined by recurring episodes of fever and serositis, usually self-limiting. It especially affects people with ancestors from the Mediterranean basin but can also be encountered in Asian and Ashkenazi Jews. The disease is a hereditary autosomal dominant syndrome classified as an autoinflammatory disorder, with marked elevations of pro-inflammatory markers such as CRP during both attacks and remission. Clinical manifestations can last a few days and usually resolve spontaneously; they include high fever and diffuse pain in the abdomen or joints. Fever is usually present during crises. Serositis such pleural and pericardial effusions and hydroceles is often present.
Since 1997, we have known that mutations in the MEFV gene, located on chromosome 16, are associated with this disease. This gene encodes the protein pyrin (marenostrin), which takes part in the inflammasome. More than 310 mutations have been described to date. Many gene variants have been linked to a pathological state, although five are still of undetermined significance (E148Q, K695R, P369S, F479L and I591T)[1].
p.Glu148Gln (PQ148) is one of the most common variants in FMF. Cases of severe FMF have been described in patients homozygous for PQ148. It is usually pathogenic when associated with other variants, such as V726A. p.Pro369Ser (P369S) is another variant but only one case has been described as pathogenic. Association of P369S with other variants such as R408Q showed variable phenotypes, although some patients had a typical FMF presentation[2, 3].
In a hereditary autosomal dominant disease like FMF, the association of different genetic variants could cause a pathogenic state via compound heterozygosity. Indeed, both parents could be healthy carriers of a single variant that could become pathological in their offspring if associated with the other variant.
To the best of our knowledge, only three other reported cases associating both p.Pro369Ser and p.Glu148Gln have been described in the literature (Table 1). The first patient reported by Battal et al. presented only with abdominal pain[3]. Another patient presented with fever, arthralgia and abdominal pain and had multiple mutations (p.Glu148Gln, p.Pro369Ser and p.Arg408Gln) in the MEFV gene[4]. The last case we found was described in 2018 and did not show any pathogenicity[5]. Our patient first presented with abdominal pain but developed serositis accompanied by fever.
Testing our patient’s parents and sibling would have been an elegant way to confirm the role of these two mutations in the pathological state, but they refused permission. The fact that attacks were occurring after treatment with colchicine had been stopped and improved when colchicine was started again was proof ex juvantibus that confirmed our clinical impression and genetic tests.
CONCLUSION
It is crucial to report symptoms caused by variants of unknown significance in order to achieve better understanding of clinical presentations and the best way to manage them. Although rarely reported, the heterozygous state of p.Pro369Ser and p.Glu148Gln, when associated, might cause typical FMF symptoms.
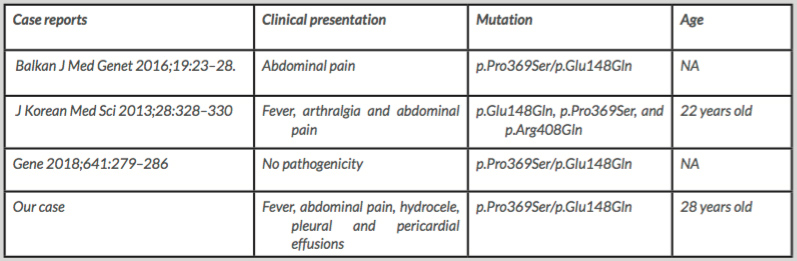
Table 1 (click to enlarge)
Table 1. Cases harbouring both p.Pro369Ser and p.Glu148Gln and their clinical characteristics