ABSTRACT
Pituitary apoplexy is a rare medico-surgical emergency that stems from an acute expansion of a pituitary adenoma from infarction or haemorrhage and where the treatment strategy is still controversial. Clinical presentation is highly variable and a high index of suspicion is needed to make the diagnosis. Furthermore, in less than half of cases, a precipitating event is identified. We report a case of a 74-year-old female who, after introduction of anticoagulation for pulmonary thromboembolism, presented with pituitary apoplexy heralded by acute adrenal insufficiency, headaches, visual symptoms and hypogonadotropic hypogonadism. Timely initiation of corticosteroids was crucial, and after stabilisation, a conservative treatment strategy was favoured with good long-term prognosis. Long-term follow-up of pituitary function also revealed new growth hormone deficiency.
LEARNING POINTS
- Corticosteroid therapy may be crucial in the emergency setting, and it is recommended for all patients with suspected pituitary apoplexy (PA).
- Early recognition of PA and its predisposing factors is crucial for the best outcome for the patient.
- Initial conservative treatment strategies are gaining popularity but close clinical monitoring is fundamental to recognise the need for sellar decompression.
KEYWORDS
Pituitary apoplexy, hypopituitarism, adrenal insufficiency, low molecular weight heparin, headache
INTRODUCTION
Pituitary apoplexy (PA) is characterised by infarction and/or haemorrhage within the pituitary gland, most commonly, in the presence of a pre-existing pituitary adenoma[1]. It is a rare disease with an estimated prevalence of 6.2 cases per 100,000 habitants[2]. The abrupt change in intrasellar pressure and compression of adjacent structures inside the rigid cavity of the sella turcica results in the classic manifestations of PA (headaches, visual impairment and ocular palsy)[3].
This case illustrates the clinical presentation and endocrinological evolution of a patient with acute PA with visual impairment and adrenal insufficiency who underwent conservative management with overall good outcomes. In addition, the importance of early recognition and consideration of precipitant factors is highlighted in this case.
CASE DESCRIPTION
A 74-year-old woman, with a history of arterial hypertension, type 2 diabetes mellitus and primary hypothyroidism, presented at the emergency department with chest pain and dyspnoea. She was diagnosed with pulmonary embolism and low molecular weight heparin (LMWH) was initiated. Four days after initiating LMWH, the patient suddenly experienced vomiting and an intense supraorbital bilateral headache associated with blurry vision. Physical examination was remarkable for bitemporal hemianopsia and blood pressure of 100/69 mmHg. There was no evidence of consciousness impairment, meningeal irritation, fever, ocular palsy nor other cranial nerve deficits. Strength and gross pain sensibility were preserved and symmetrical.
An urgent CT scan of the brain revealed a haemorrhagic lesion in the pituitary gland, establishing contact with the optic chiasm. Anticoagulation was suspended and intravenous fluid replacement and corticosteroids were initiated. Blood samples showed mild hyponatraemia and determination of hypothalamic–pituitary axis hormone levels showed an isolated hypogonadotropic hypogonadism (Table 1). Cortisol levels were not assessed as steroid therapy had already been started. Computerised perimetry (CP) on the third day after symptom onset confirmed bitemporal hemianopsia. Magnetic resonance imaging (MRI) showed an intra- and suprasellar expansive lesion (24×20 mm) that protruded against the optic chiasm (Fig. 1).
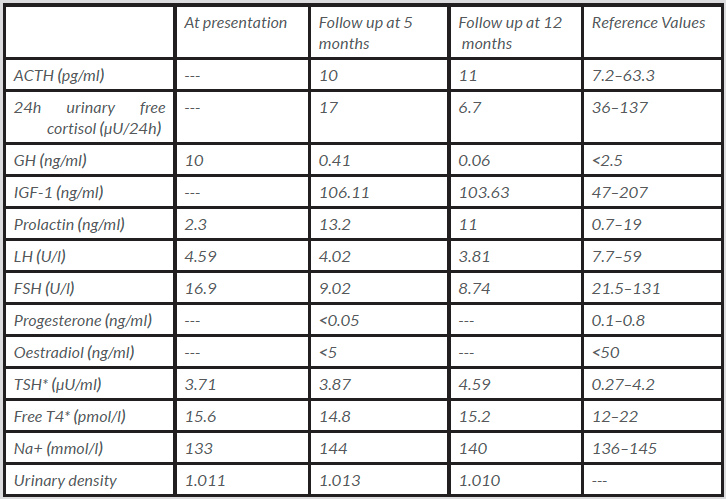
Table 1 (click to enlarge)
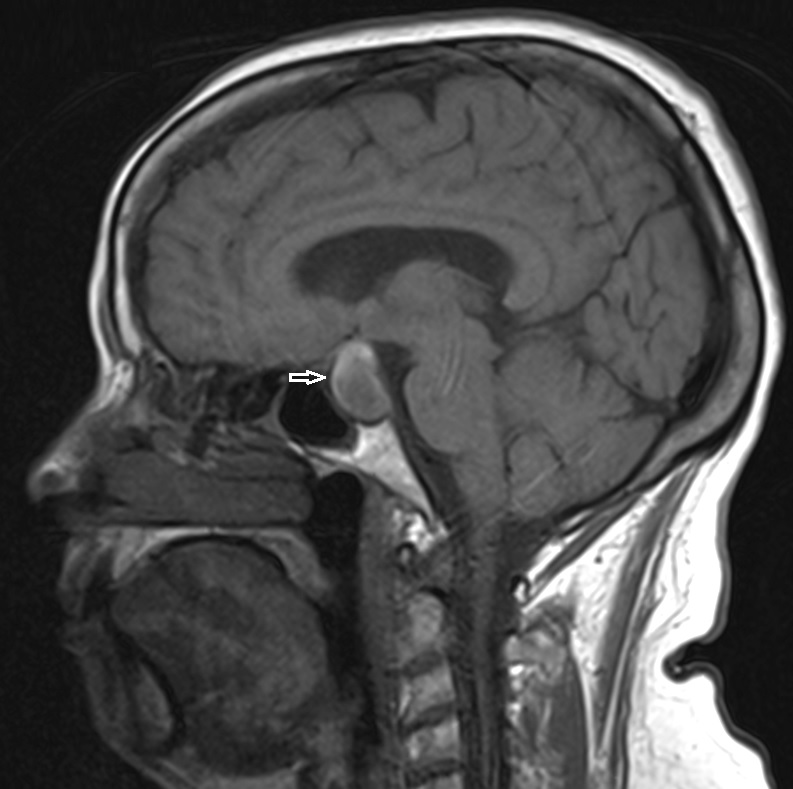
Figure 1 (click to enlarge)
Table 1. Laboratory results
*On levothyroxine 0.025 mg/day.
ACTH: adrenocorticotropin; FSH: follicle-stimulating hormone; LH: luteinising hormone; IGF-1: insulin-like growth factor 1; TSH: thyroid-stimulating hormone; T4: thyroxine.
Figure 1. Intrasellar and suprasellar expansive lesion with haemorrhage
The patient experienced a favourable clinical course with resolution of headaches and partial improvement in visual field impairment during the first week, which supported a non-surgical treatment approach.
She was discharged on prednisolone 5 mg/day. Follow-up was unremarkable with consistent improvement in visual acuity confirmed by repeated CP. An MRI scan performed at 6 months showed marked reduction of the lesion (6×15 mm) without optic chiasm compression. At the 1-year follow-up, the patient remained asymptomatic, with persistence of hypogonadotropic hypogonadism, with added growth hormone deficiency and borderline adrenal insufficiency after steroid tapering (Table 1).
DISCUSSION
PPA complicates 2–12% of patients with a pituitary adenoma, with non-functioning macroadenomas being the most frequent subtype[3]. Precipitant factors are recognised in 10 to 40% of cases, in which antithrombotic therapy emerges as an important one of these factors[3,4]. In this case, early diagnosis was crucial, allowing for prompt discontinuation of anticoagulation and facilitating the recognition of adrenal insufficiency in a timely manner.
A high index of suspicion is necessary for the diagnosis because symptoms at presentation can be highly variable, and often subclinical. However, in its most severe form, PA can be a true medical and neurosurgical emergency with risk of permanent blindness and haemodynamic instability.
Treatment of PA is focused on medical management and sellar decompression if warranted. Acute medical management is focussed mainly on mitigating adrenal insufficiency (heralded by haemodynamic instability, hypoglycaemia and hyponatraemia). As the estimated prevalence of adrenal insufficiency in PA is 50–80%, corticosteroid therapy should be initiated in all patients with confirmed or suspected PA[3]. In this case, evidence for adrenal insufficiency was related to relative hypotension (especially in a previously hypertensive patient) and newly developed hyponatraemia. Whenever possible, evaluation of the adrenocorticotropic axis should be performed before administration of steroids to correctly interpret the results. Other pituitary axis hormones are also frequently affected, although they are not usually prominent within the acute clinical picture.
Chronic medical therapy is related to the severity of pituitary deficiency. In the long-term, endocrine function often remains altered and seems independent from the treatment strategy adopted[3]. Growth hormone deficiency is present in almost all patients, hypocortisolism in 50–80%, gonadotrophin insufficiency in 40–75%, central hypothyroidism in 30–70% and a prolactin deficit is seen in 10–40% of cases[3]. Still, data regarding the long-term evolution of pituitary abnormalities is scarce. Interestingly, in our case, growth hormone deficiency was not present at presentation.
Even though in the past, PA was treated almost exclusively neurosurgically, conservative treatment has gained popularity in the last decade[3]. However, to date, there are still no evidence-based standards for optimum care regarding different treatment strategies given the retrospective nature of published studies that suffered from a selection bias (surgical patients had a more severe presentation)[5]. According to the United Kingdom guidelines (2010), indications for surgery are: severe neuro-ophthalmic signs (severely reduced visual acuity); severe and persistent or deteriorating visual field defects; and a deteriorating level of consciousness[5]. However, the definition of severe visual impairment is vague and must be adjudicated on an individual basis.
Non-severe visual impairments tend to progress favourably with a good long-term prognosis. Ocular palsies usually resolve within weeks with conservative treatment, and as such they are not determinants in treatment decision-making[3]. Our case represents a clear example where a conservative strategy with close monitoring yielded favourable results.
CONCLUSION
This case highlights important messages regarding management of PA that may influence outcomes: early diagnosis, consideration of precipitant factors that may be reversed and initiation of steroids may be crucial in an emergency setting. Deciding on the best treatment strategy depends on the state of consciousness and neuro-ophthalmologic impairment, and should be performed by an experienced multidisciplinary team, as few quality studies exist with respect to deciding on the best candidates for surgical decompression.