ABSTRACT
Tuberous sclerosis complex (TSC) is a rare, autosomal dominant disorder with a recognized phenotypic variability with clinical manifestations developing continuously throughout life. The follow-up of TSC patients is challenging. The authors present a case with a definite diagnosis of TSC with known skin, renal, hepatic and neuropsychiatric involvement, whose diagnosis of TSC-associated lymphangioleiomyomatosis was establish at a late stage after the patient had presented with recurrent pneumothorax. Notwithstanding, mammalian target of rapamycin inhibition therapy was initiated.
LEARNING POINTS
- Tuberous sclerosis complex (TSC) is a systemic and progressive disorder leading to significant morbidity, highlighting the need to closely monitor such patients.
- TSC-associated lymphangioleiomyomatosis is the major pulmonary manifestation and screening at an early stage is imperative.
- Managing TSC requires a multidisciplinary approach and the introduction of mammalian target of rapamycin (mTOR) inhibition therapy reinforces the need for collaboration between well-prepared health professionals.
KEYWORDS
Tuberous sclerosis complex, lymphangioleiomyomatosis, pneumothorax
INTRODUCTION
Tuberous sclerosis complex (TSC) is a rare, autosomal dominant neurocutaneous disorder with an incidence of approximately 1 in 6,000–10,000 live births[1]. Mutations in the TSC1 and TSC2 tumour suppressor genes occur in the majority of patients, resulting in hyperactivation of the mammalian target of rapamycin (mTOR) signalling pathway and subsequent abnormalities in cell differentiation and proliferation with the growth of hamartomatous and dysplastic lesions affecting multiple organs including the brain, heart, kidneys, lungs and skin[2]. Although it has nearly complete penetrance, this disorder has a variable expression, so the symptoms and severity of TSC differ widely between individuals and there is great phenotypic variability[2]. The authors present a rare case of TSC-associated lymphangioleiomyomatosis (TSC-LAM), a cystic interstitial lung disease representing a major manifestation of pulmonary involvement by the disease.
CASE DESCRIPTION
The patient was a 35-year-old woman with a history of tuberous sclerosis with documented neuropsychiatric (childhood epilepsy and intellectual disability), cutaneous (facial angiofibroma and a shagreen patch) (Fig. 1), renal and hepatic (angiomyolipomas, AML) involvement. At the age of 18 she had been submitted to right nephrectomy due to AML rupture. She had stable chronic kidney disease (CKD; stage 3) and multifactorial anaemia secondary to CKD and iron deficiency (menorrhagia) and was medicated with iron, vitamin D and progestational supplements. She was a non-smoker.
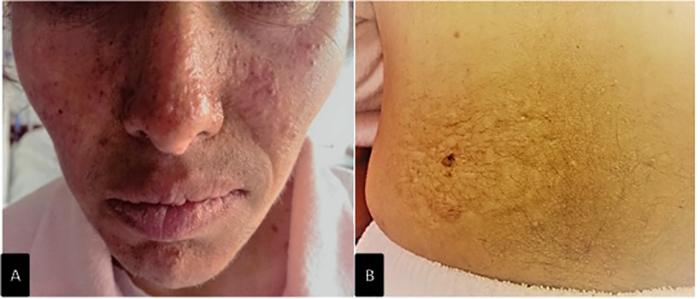
Figure 1 (click to enlarge)
Figure 1. (A) Facial angiofibroma; (B) shagreen patch
The patient presented with sudden right-sided chest pain and dyspnoea. She had a history of a productive cough persisting for over 2 weeks. At evaluation, tachypnoea, tachycardia and absence of breath sounds throughout the right chest field were observed. Hypoxaemia was documented (on room air: pCO2 29 mmHg, pO2 49 mmHg) which responded well to oxygen supplementation. There were no other significant findings.
Chest radiography confirmed the clinical suspicion of pneumothorax, without signs of tension, and showed bilateral parenchymal areas compatible with pulmonary cysts (Fig. 2). Thoracic drainage was performed and the patient was admitted to the admitted to a general ward (follow-up radiography showed improvement of the pneumothorax). The patient’s symptoms fully resolved following lung re-expansion. High-resolution chest CT (chest HRCT) revealed multiple cystic lung lesions, without sparing segments, consistent with late stage lymphangioleiomyomatosis (LAM) (Fig. 3), confirming the pulmonary involvement of the TSC. Pulmonary function tests (PFT) revealed a severely reduced diffusing capacity of the lung for carbon monoxide (27%), with severe obstructive ventilatory syndrome with reduced forced expiratory volume in 1 second and reduced ratio of FEV1 to forced vital capacity. At discharge the patient was on bronchodilators.
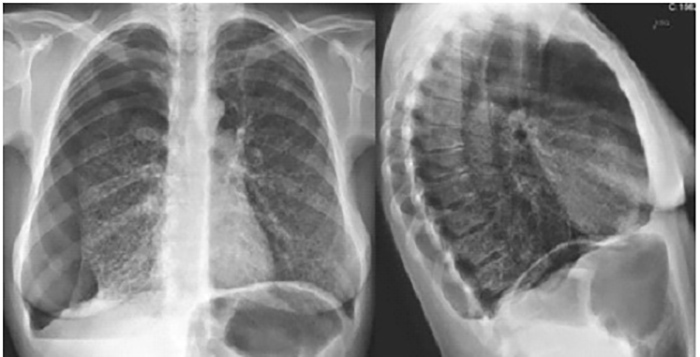
Figure 2 (click to enlarge)
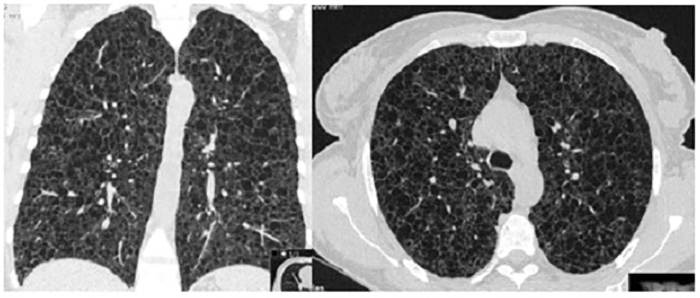
Figure 3 (click to enlarge)
Figure 2. Chest radiography at admission in the emergency department
Figure 3. High resolution chest computed tomography
DISCUSSION
TSC can be diagnosed through genetic study or by its clinical manifestations. Genetic diagnosis is based on identification of a mutation that clearly prevents protein synthesis and/or implies impaired function of the TSC1 or TSC2 proteins[2]. The clinical diagnostic criteria for TSC were reviewed at the second International Tuberous Sclerosis Complex Consensus Conference in 2012. The criteria include 11 ‘major’ and 6 ‘minor’ features indicating either definite or possible TSC, respectively[1,2].
Our patient had a previous definite diagnosis of TSC but no pulmonary involvement had been documented. The follow-up of TSC patients is well established and concerns about multisystemic surveillance have been highlighted[3] All TSC patients are at risk for TSC-LAM, especially women aged 18 years or over, and should undergo a baseline chest HRCT. Symptoms should be enquired about at each clinical examination and in asymptomatic individuals with a baseline chest HRCT without lung cysts, imaging must be repeated every 5–10 years; if cysts are detected, the gap should be reduced to 2–3 years and an annual PFT and a 6-minute walk test to determine the rate of progression should also be included. If there is evidence of severe TSC-LAM, PFT and a chest HRCT may be needed as frequently as every 3–6 months to guide treatment decisions[3]. LAM has three major presentations: dyspnoea on exertion, spontaneous pneumothorax or incidental discovery on imaging. In contrast to the sporadic form of the disease, TSC-LAM is diagnosed at an earlier stage due to active case screening[4].
We had no access to or knowledge of previous examinations or pulmonary history and unfortunately this patient presented with severe lung disease. As described in the literature, the degree of impairment on PFT correlates with the severity of abnormalities seen on HRCT. After complete evaluation, the patient was referred for specific consultation and treatment with a mTOR inhibitor was initiated. mTOR inhibitors such as sirolimus and everolimus have the potential to provide targeted therapy for patients with TSC and are approved for four distinct manifestations of the disease: giant cell astrocytoma, angiomyolipoma, lymphangioleiomyomatosis and epilepsy[5]. Sirolimus is generally favoured for TSC-LAM because of the lack of randomized studies of everolimus for this condition, but it is reasonable to assume that both drugs will have similar beneficial treatment effects[4]. The prognosis of TSC-LAM patients is poor, with previous descriptions of death secondary to respiratory insufficiency often within 5 years of the onset of symptoms. Nowadays, long-term survival may be more common because of improved management of the disease and its potential complications (especially cor pulmonale and pneumothorax)[4].
CONCLUSION
The authors present a typical case demonstrating the multisystemic involvement of TSC, a disease that requires multidisciplinary surveillance because of its recognized clinical pleiotropy. The pulmonary involvement with recurrent pneumothorax and decreased ventilatory function in our patient is highlighted. LAM is the major form of pulmonary involvement in patients with TSC, and accounts for a significant proportion of morbidity and mortality. HRCT and PFT should be considered at baseline and in the follow-up of these patients because early detection of abnormalities may require attention and/or specific therapy in order to prevent disease progression and poor outcomes.