ABSTRACT
Lysosomal storage disorders (LSDs) are a group of genetic disorders caused by mutations in genes encoding enzymes involved in lysosomal function. Schindler disease is an autosomal recessive, inherited LSD caused by defective or non-existent activity of the enzyme α-N-acetylgalactosaminidase (α-NAGA). To date, three main phenotypes of Schindler disease have been described. We report the case of a 68-year-old man presenting with axonal and demyelinating polyneuropathy, sensorineural hearing loss, chronic lymphoedema, angiokeratoma corporis diffusum and bilateral carpal tunnel syndrome. Genetic testing (PCR) for α-galactosidase revealed the c.577G>T (p.Glu193*) mutation in the NAGA gene, confirming Schindler disease, which is clinically compatible with Kanzaki disease and Schindler disease type II.
LEARNING POINTS
- Schindler disease is a very rare lysosomal storage disorder.
- To our knowledge, fewer than 20 cases have been described to date.
- Consequently, each new case should be reported to enhance understanding of the wide range of presentations.
KEYWORDS
Angiokeratoma corporis diffusum, Kanzaki disease, peripheral neuropathy, alpha-N-acetylgalactosaminidase
INTRODUCTION
Lysosomal storage disorders (LSDs) are a group of genetic disorders caused by mutations in genes encoding enzymes involved in lysosomal function, leading to their respective substrates accumulating within lysosomes. Schindler disease is an autosomal recessive, inherited LSD caused by defective or non-existent activity of the α-N-acetylgalactosaminidase enzyme (α-NAGA), resulting in mild to severe clinical features. α-NAGA, also known as galactosidase B, shares some sequence identity with the GLA gene, encoding for α-galactosidase, the enzyme deficient in Fabry disease. To date, three main phenotypes of Schindler disease have been described (I, II, III), ranging from an infantile-onset neuroaxonal dystrophy to an adult-onset disorder with mild intellectual disability. The only treatment for this disease consists of support measures and symptomatic treatment, although some other therapies have been proposed.
CASE REPORT
Our patient is a 68-year-old man presenting with axonal and demyelinating polyneuropathy (causing neuropathic pain), sensorineural hearing loss, chronic lymphoedema, angiokeratoma corporis diffusum and bilateral carpal tunnel syndrome. His only treatment is simple analgesia on demand for his polyneuropathy.
Neither of his healthy parents have any distinguishing phenotypical features. He is the fourth of five children: a deceased sister showed some features of the condition, but none of his brothers have known disease.
On physical examination, the patient had a coarse facies and bilateral symmetrical massive lymphoedema. His skin had a thickened nodular surface with brownish pigmentation (Fig. 1a). Multiple millimetre-sized back-reddish papules could be seen (Fig. 1b) on the abdomen, groin and inner aspects of both arms, which dermatoscopy revealed as red lacunae (Fig. 1c).
Histopathological examination revealed angiokeratomas underlying superficial (upper dermis) vascular ectasia (Fig. 2a) and epidermal hyperplasia (acanthosis and/or hyperkeratosis) (Fig. 2b). Echocardiography (moderate cardiomegaly), chest x-ray, EKG and urine tests (with urine sediment) showed results within normal ranges. The patient is blood group A. His intelligence quotient (IQ) is slightly lower than normal, with normal verbal IQ but slightly reduced performance.
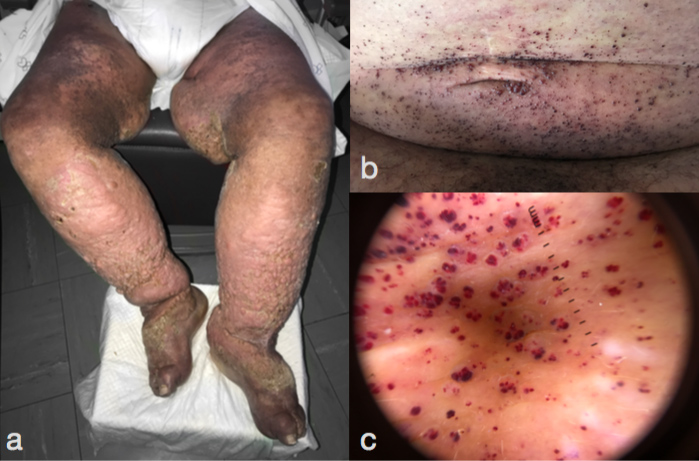
Figure 1 (click to enlarge)
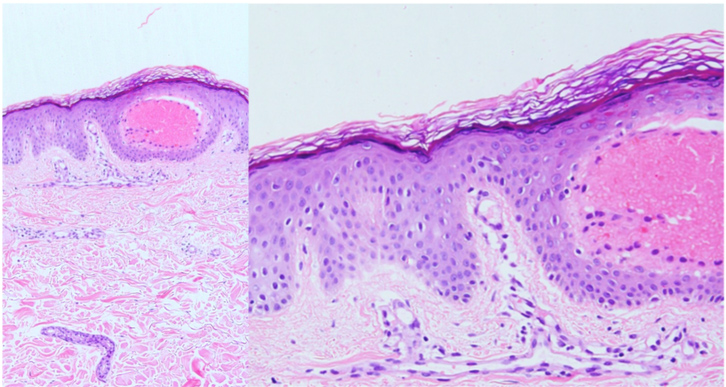
Figure 2 (click to enlarge)
Figure 1. Symmetrical massive lymphoedema, with thickened nodular skin and brownish pigmentation. (a) On the abdomen, multiple angiokeratomas (b) can be seen, which under dermatoscopy show red lacunae (c)
Figure 2. Angiokeratomas with superficial (upper dermis) vascular ectasia (a) and overlying epidermal hyperplasia (acanthosis and/or hyperkeratosis) (b)
DISCUSSION
Schindler disease is an autosomal recessive, inherited LSD caused by defective or non-existent activity of the enzyme α-NAGA[1]. The defective functioning of this enzyme (and thus the blocking of hydrolysis of terminal, non-reducing α-D-galactose residues in α-galactosides) results in large molecules such as galactose oligosaccharides, galactomannans and galactolipids accumulating within cells, resulting in a wide phenotypic spectrum. Given the ubiquitous character of this enzyme and of oligosaccharides, signs and symptoms may be systemic.
Since the first case was reported in 1987, three main phenotypes have been described: type I (infantile-onset), type II (adult-onset) and type III (intermediate phenotype). Schindler disease type I generally develops around the second year of life, presenting with hypotonia, areflexia, rigidity, spasticity, deafness, blindness and mental deterioration. This typically results in death before the age of 6 years. Schindler disease type II (also known as Kanzaki disease) is characterised by lymphoedema, angiokeratoma corporis diffusum, neurological involvement, peripheral neuropathy, sensorineural hearing loss and recurrent vertigo attacks (with doubtful association with Ménière’s disease). Cardiac involvement in the form of moderate cardiomegaly or hypertrophy of the interventricular septum can also occur[2]. Finally, Schindler disease type III shows an intermediate phenotype between types I and II[1].
Patients with blood group A have a worse prognosis. As the terminal residue of blood group antigen A, which should be cleaved by α-NAGA, is deficient in patients with Schindler disease, this results in more severe storage disease, as in our patient[1,3].
Clinical diagnosis is difficult and usually delayed for years. Enzymatic analysis through a blood test shows diminished α-NAGA activity. In addition, urine tests can identify glycopeptiduria, as in Fabry disease. However, genetics testing (PCR) is the gold-standard method for diagnosis.
There is no current treatment for Schindler disease, although two pharmacological chaperones (which are proteins that help the assembly and disassembly of macromolecular structures) have been proposed as potential therapeutic agents[1].
Our patient was clinically and biochemically diagnosed[3] as having Kanzaki disease in 1994. In 1993 and 2010, enzymatic analysis for Fabry disease conducted in peripheral blood fibroblasts and leucocytes showed α-galactosidase activity was normal. However, genetic testing (PCR) for α-galactosidase in 2010 accidentally revealed an apparently homozygous c.577G>T (p.Glu193*) mutation in the NAGA gene, thus confirming Schindler disease. As a result, genetic testing was conducted for all surviving family members, with no mutations found[3]. Currently, our patient’s polyneuropathy, hearing loss and angiokeratomas is clinically stable, but his chronic lymphoedema is slowly progressing.
- Ferreira CR, Gahl WA. Lysosomal storage diseases. Transl Sci Rare Dis 2017;2:1–71.
- Kodama K, Kanzaki T, Abe R, Ohkawara A, Yoshii N, Yotsumoto S, et al. A new case of alpha-N-acetylgalactosaminidase deficiency with angiokeratoma corporis diffusum, with Ménière's syndrome and without mental retardation. Br J Dermatol 2001;144:363–368.
- Chabás A, Coll MJ, Aparicio M, Rodriguez Diaz E. Mild phenotypic expression of alpha-N-acetylgalactosaminidase deficiency in two adult siblings. J Inherit Metab Dis 1994;17:724–731.