ABSTRACT
Whipple's disease is a rare multisystemic infectious disease that can mimic lymphoproliferative disorders and must be considered in the differential diagnosis of febrile syndromes. The authors describe the case of a 55-year-old man who presented to the Emergency Department with dyspnoea and abdominal pain. He had a 2-month history of fever, night sweats, asthenia and unintentional weight loss. Upon clinical examination he had bilateral inguinal lymphadenopathy. Blood tests showed iron-deficit anaemia and high C-reactive protein. Abdominal ultrasound showed mesenteric and iliac adenopathies and hepatosplenomegaly. The patient was admitted to the Internal Medicine department for additional testing. Flow cytometry analysis of peripheral blood showed CD5-positive monoclonal B-cell expansion. Excisional biopsy of a retroperitoneal adenopathy guided by computed tomography showed periodic acid–Schiff-positive bacilli inside the macrophages, further identified as Tropheryma whipplei through polymerase chain reaction. Bone marrow biopsy showed a scarce positive CD5 lymphoid population and haematopoietic alterations related to infection. The patient started treatment for T. whipplei with complete symptom resolution. This is the first case describing the simultaneous diagnosis of Whipple's disease and chronic lymphocytic leukaemia in a patient with constitutional symptoms, fever and lymphadenopathies.
LEARNING POINTS
- Whipple's disease can mimic lymphoproliferative disorders and should be considered in the differential diagnosis of lymphadenopathy and fever, even in the absence of gastrointestinal symptoms.
- Most patients with chronic lymphocytic leukaemia are asymptomatic, but a minority have B symptoms and increased risk of infections.
- An accurate clinical history and differential diagnosis are fundamental; if the authors had not had a high level of suspicion, this patient could have been diagnosed with just chronic lymphocytic leukaemia and may have died if Whipple's disease had been left untreated.
KEYWORDS
Fever, lymphadenopathy, Whipple's disease, chronic lymphocytic leukaemia, small lymphocytic lymphoma
CASE REPORT
A 55-year-old Caucasian man presented to the Emergency Department with dyspnoea and abdominal pain, without other gastrointestinal or respiratory symptoms. He also had a 2-month history of fever, night sweats, asthenia and unintentional weight loss (10% of basal weight). Upon physical examination he had bilateral inguinal lymphadenopathy, without other alterations.
He had a previous medical history of obesity and obstructive sleep apnoea. He was taking no medication. He was an ex-smoker, with minimum alcohol intake and did not have any recent travel history. He lived in a rural area and worked as a house-builder.
Blood tests showed microcytic hypochromic anaemia with iron deficit (haemoglobin 10.7 g/dl, normal value 13–18 g/dl; mean corpuscular volume 75.1 fl, normal value 87–103 fl; mean corpuscular haemoglobin 25.7 pg, normal value 27–35 pg; 31.3% haematocrit, normal value 43–55%; transferrin saturation of 9%, normal value 20–50%), high C-reactive protein (244 mg/l, normal value <3 mg/l) and high sedimentation rate (130 mm/h, normal value 0–15 mm/h), without leucocytosis, neutrophilia or lymphocytosis. Chest radiography and arterial blood gas testing were normal. Abdominal ultrasound showed widespread mesenteric and iliac adenomegalies and hepatosplenomegaly. The patient was admitted to the Internal Medicine department for additional testing.
METHODS AND PROCEDURES
CT of the chest, abdomen and pelvis confirmed widespread retroperitoneal, mesenteric and iliac adenomegalies and hepatosplenomegaly (Fig. 1).
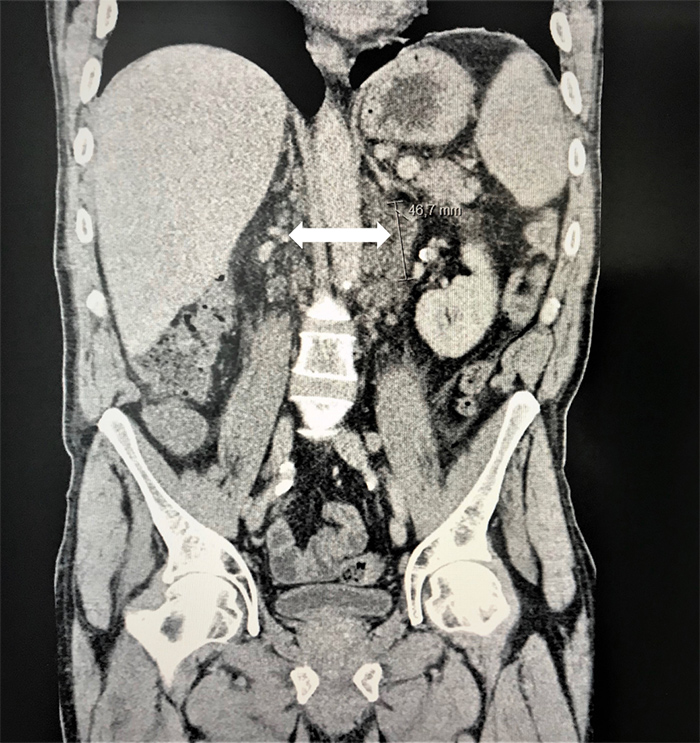
Figure 1 (click to enlarge)
Figure 1. Computed tomography of the abdomen and pelvis confirmed widespread retroperitoneal, mesenteric and iliac adenomegalies and hepatosplenomegaly. The arrow points to the largest conglomerate of lymphadenopathies
Human immunodeficiency virus testing was negative. Blood cultures and the Mycobacterium tuberculosis direct test in respiratory samples were negative. An angiotensin-converting enzyme assay was normal. Immunological blood tests were normal, except for a slight elevation in immunoglobulin G4 (220 mg/dl, normal value 40–130 mg/dl). Gastroduodenal endoscopy was performed with duodenal biopsies, as was total colonoscopy; histology was normal. Fine-needle aspiration of an inguinal lymph node was inconclusive. Flow cytometry analysis of blood showed a positive CD5 monoclonal B-cell expansion. An excisional biopsy of a retroperitoneal adenopathy guided by CT was requested and showed periodic acid–Schiff (PAS)-positive bacilli inside the macrophages, identified as Tropheryma whipplei in polymerase chain reaction (PCR) (Fig. 2). Bone marrow biopsy showed a scarce positive CD5 lymphoid population and haematopoietic alterations reactive to infection.
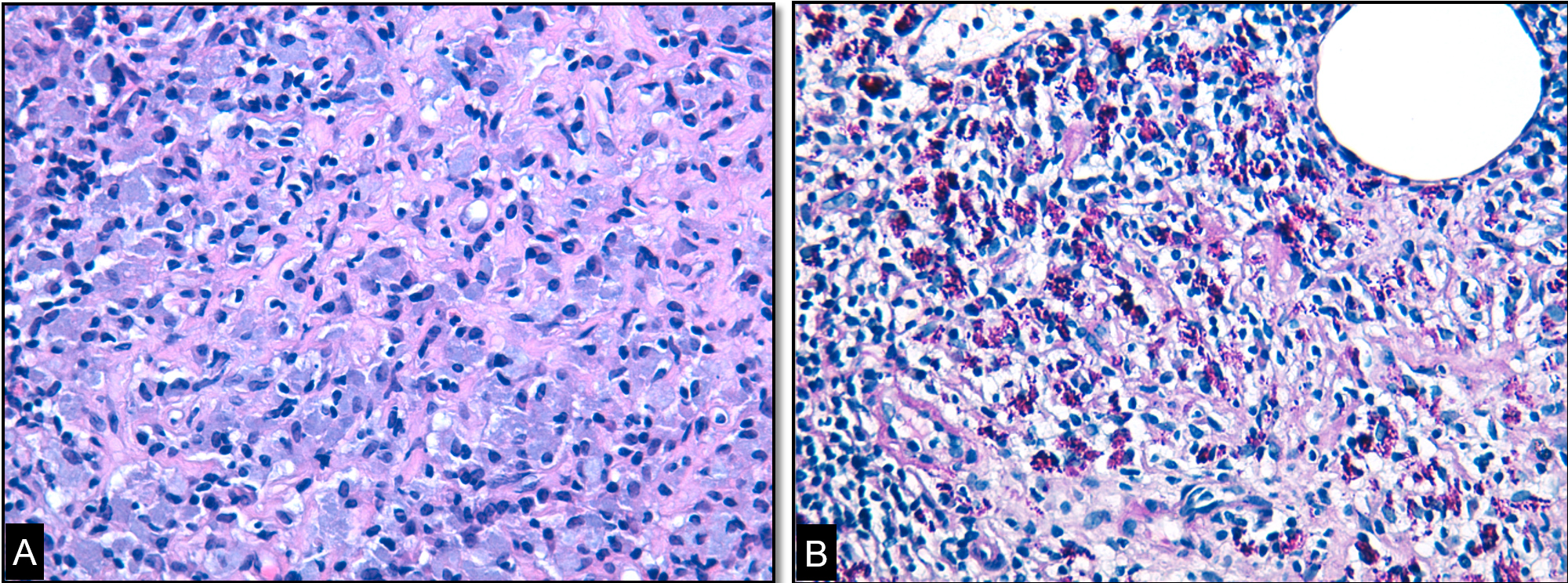
Figure 2 (click to enlarge)
Figure 2. (A) Lymph node microbiopsy where numerous histiocytic cells are identified, in a ‘twisted towel’ arrangement, with abundant basophilic cytoplasm.(B) In the histochemical study with periodic acid–Schiff after diastasis, structures compatible with Tropheryma whipplei are observed in the histiocyte cytoplasm. Note the presence of a lipid vacuole typical of this entity
The patient started a 2-week antibiotic course of ceftriaxone, switched afterwards to an oral regimen of trimethoprim-sulfamethoxazole (160/800 mg). Within 2 months of a planned year-long treatment regimen, the patient had complete resolution of symptoms and had gained 5 kg.
DISCUSSION
Whipple's disease (WD) is a rare multisystemic infectious disease caused by the Gram-positive bacillus T. whipplei. WD can mimic lymphoproliferative disorders, malabsorption syndromes or rheumatic diseases[1,2]. The clinical spectrum of WD includes four types of manifestations: the classic form with chronic diarrhoea, abdominal pain, weight loss and arthralgias; the localized type, including endocarditis, uveitis, lymphadenitis and neurological manifestations; acute self-limited infections, such as acute gastroenteritis or pneumonia; and finally, the asymptomatic healthy carriers[3]. The prevalence of mesenteric lymphadenopathy in Whipple’s disease is reported to be 17% [2].
WD can be fatal if not correctly diagnosed and treated, and a high level of suspicion is required for diagnosis[1,2]. The incidence is unknown; patients are typically Caucasians males, with a mean age of 50 years and a history of agricultural exposure, as in our patient[1].
The gold standard diagnostic test used to be PAS staining of duodenal biopsy specimens, but this has poor specificity and sensitivity[3]. PCR has led to an expansion in diagnostic capacity, especially in localized WD with lymphadenitis, as reported in this case[1].
Patients diagnosed with WD should be treated with 2 weeks of parental antibiotics that can cross the blood–brain barrier, irrespective of suspected neurological involvement, followed by at least a year of oral antibiotics[1].
Chronic lymphocytic leukaemia (CLL) or small lymphocytic lymphoma (SLL) are two manifestations of the same disease, a mature B-cell neoplasm characterized by progressive accumulation of monoclonal B-lymphocytes. The term CLL is used when the disease manifests primarily in the blood, whereas SLL is used when involvement is primarily lymph-nodal. Most patients are asymptomatic, but a minority have B symptoms and increased susceptibility to infections. Our patient became asymptomatic after starting WD-directed treatment. After 5 years, he maintains only clinical vigilance since CLL does not require treatment unless symptoms or severe cytopenia develop[4].
In the literature, there is only one reported case of an association between these two entities, in which WD appears as a complication of CLL after 6 years of treatment with fludarabine[5].