ABSTRACT
A 43-year-old Caucasian male initiated myalgias and loss of muscle strength in the upper and lower limbs, but especially at the shoulder and pelvic girdle. Creatinine phosphokinase was elevated seven-fold above the normal reference value and aldolase was slightly elevated. He had a previous diagnosis of Behçet's disease, antiphospholipid syndrome and hypertriglyceridaemia. At this time, he was on azathioprine 150 mg daily, colchicine 1 mg daily, warfarin and fenofibrate 200 mg daily. Fenofibrate was stopped and creatinine phosphokinase re-evaluated 2 months later, but it was higher, with persistent myalgias. By this time, prednisolone was restarted and the azathioprine dose reduced until it was discontinued. Nevertheless, 2 months after stopping azathioprine, the patient remained symptomatic and creatinine phosphokinase was persistently elevated. At this point, the authors requested myositis antibody testing to exclude overlap with a third autoimmune disorder, and Ro52 antibody was positive. Electromyography was normal. Magnetic resonance imaging of lower limb muscles was compatible with polymyositis. Muscular biopsy of the medial gastrocnemius revealed inflammatory myopathy. The authors proposed treatment with rituximab and after 3 months, the patient had clinically and analytically improved, with reduction of creatinine phosphokinase, without adverse reactions. As we can see in this case, rituximab could be a secure treatment for patients with idiopathic inflammatory myopathy without improvement on glucocorticoids plus another immunosuppressive agent. This patient has a rare overlap syndrome, since this is the first case of an association between inflammatory myopathy, Behçet's disease and antiphospholipid syndrome described in the literature.
LEARNING POINTS
- This is the first case report in the literature of an association between inflammatory myopathy, Behçet's disease and antiphospholipid syndrome.
- Rituximab could be a secure treatment for refractory idiopathic inflammatory myopathy.
- This case report highlights the importance of a methodical diagnostic work-up for an accurate diagnosis.
KEYWORDS
Rituximab, inflammatory myopathy, polymyositis, Behçet's disease, antiphospholipid syndrome
CASE DESCRIPTION
A 43-year-old Caucasian male initiated myalgias and symmetric loss of muscle strength in the upper and lower limbs, but especially at the shoulder and pelvic girdle. Creatinine phosphokinase (CK) was elevated (713 U/l, normal reference 10–172 U/l) as well as aldolase (8.9 U/l, normal value <7.6) and hepatic enzymes (aspartate transaminase 43 U/l and alanine transaminase 65 U/l, normal reference 10–37 U/l). Upon physical examination, no alterations were found beyond grade 4/5 strength in the upper and lower limbs, predominantly proximal. Calcinosis, arthritis, heliotropic rash, Gottron’s papules, cutaneous nodules and erythema were absent. The patient had a medical history of Behçet's disease (BD) since he was 30 years old; antiphospholipid syndrome (APS) diagnosed 7 years previously after left iliofemoral vein thrombosis; and hypertriglyceridaemia. He was an ex-smoker, with minimal alcohol intake and did not report any recent travel history. He worked as a postman. At this time, he was medicated with azathioprine (AZA) 150 mg daily, colchicine 1 mg daily, warfarin and fenofibrate 200 mg daily.
METHODS AND PROCEDURES
First, the authors decided to exclude iatrogenic causes of muscular symptoms and elevation of CK. The authors stopped fenofibrate and re-evaluated CK after 2 months, but the value was higher (953 U/l) and intense myalgias persisted. The authors then decided to restart prednisolone 0.5 mg/kg/daily and reduce the dose of AZA until it was fully discontinued, but 2 months after stopping AZA the patient was still symptomatic and CK was still elevated (572 U/l).
At this point, the authors requested myositis- and non-myositis-associated antibody testing to exclude overlap with a third autoimmune disorder Ro52 antibody was positive, but the other antibodies were negative , except for Lupus anticoagulant which we knew to be positive since the diagnosis of APS. (Tables 1 and 2). Electromyography (EMG) was normal. Magnetic resonance imaging (MRI) of lower limb muscles showed signs compatible with polymyositis (Fig. 1). Muscular biopsy of the medial gastrocnemius revealed signs of inflammatory myopathy.
The patient was started on rituximab (RTX) 1,000 mg (2 administrations of 15 days apart), and after 3 months, he experienced clinical and analytical improvement with reduction of CK (263 U/l).
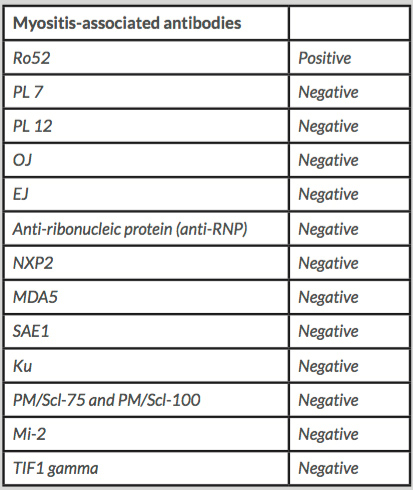
Table 1 (click to enlarge)
Table 1 (click to enlarge)
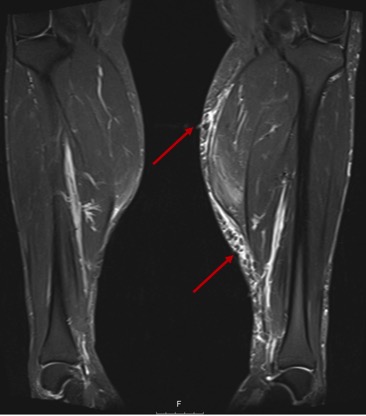
Figure 1 (click to enlarge)
Table 1. Laboratory results for myositis-associated antibodies
Table 2. Laboratory results for other antibodies
Figure 1. Magnetic resonance imaging of lower limb muscles showed muscle oedema and fatty infiltration in the superficial posterior compartment of the legs, mainly affecting the gastrocnemius bilaterally (with left predominance)
DISCUSSION
Idiopathic inflammatory myopathies (IIM) are a heterogeneous group of chronic systemic autoimmune diseases. Excluding inclusion body myositis, IIM typically present with proximal muscle weakness and may progress with systemic involvement with significant morbidity [1,2]. Serum CK is particularly important for monitoring disease progression, as well as myoglobin and aldolase. Hepatic cytolysis enzymes may be elevated secondary to both liver disease or muscle breakdown[1].
EMG has been used to distinguish neuropathic versus myopathic causes of proximal muscle weakness. MRI is gaining increased diagnostic importance, particularly for guiding muscle biopsy location. Muscle biopsy remains the gold standard for diagnosis and differentiation of subcategories of IIM[1].
Glucocorticoid therapy remains the prime first-line treatment. Refractory myopathy does not have a consensual definition, but refers to intolerance or inadequate response to glucocorticoids and at least one other immunosuppressive agent[1,2]. Multiple immunosuppressant agents have been tried as steroid-sparing agents; methotrexate along with AZA is considered first-line. Intravenous immunoglobulin G therapy is used in cases of severe or rapidly progressive disease[1–3].
This patient had initiated myositis under 1.5 mg/kg/day of AZA. This immunosuppressive agent was started 2 years previously and had been given as a full dose for over a year. The patient started colchicine due to joint involvement of BD, but without symptomatic improvement. Several months later, methotrexate was initiated, but without clinical response and the patient developed hepatotoxicity. At this time, the patient started AZA. Since myositis started under AZA and the patient had a previous history of methotrexate toxicity, these 2 drugs were considered inappropriate in this case.
Cyclophosphamide and mycophenolate mofetil had been reported in just a small retrospective series of 6 to 17 patients, particularly in patients with concomitant interstitial lung disease. Data regarding the use of calcineurin inhibitors and abatacept are limited. Several studies suggested RTX efficacy in refractory IIM with response rates ranging from 61 to 83%. In the largest trial of RTX in inflammatory myositis involving 195 patients, 83% had a clinical response and the mean dose of prednisone was significantly reduced. There are no solid data on other biologicals [2,3]. According to these data, the authors considered RTX the most appropriate treatment for this patient.
Our patient has a rare overlap syndrome, since, as far as the authors know, this is the first case report in the literature of an association between inflammatory myopathy, BD and APS. However, there are cases in the literature reporting an association between BD and APS and APS and polymyositis[4,5].