ABSTRACT
In the present report, we describe our experience with a 44-year-old male with abnormal retroperitoneal primitive neuroectodermal tumours (PNETs) in our hospital, who was operated on with a spindle cell neoplasm diagnosis.
LEARNING POINTS
- Appropriate treatment is a crucial challenge in patients with PNETs due to late referral.
- The differential diagnoses were malignant pheochromocytoma, paraganglioma and retroperitoneal sarcoma.
- Physicians should keep in mind that the patient could be simultaneously suffering from sarcoma and a retroperitoneal PNET.
KEYWORDS
Neuroectodermal tumours, male, hospital, diagnosis
INTRODUCTION
Primitive neuroectodermal tumours (PNETs) are remarkably uncommon malignant tumours originating from primitive undifferentiated small round cells at the neuroectoderm layer. PNETs are classified into central and peripheral types, based on the originating ectodermal line[1, 2]. Peripheral PNETs (pPNETs) are rare malignancies, accounting for approximately 1% of all sarcomas [3]. Although pPNETs with late manifestations and diagnosis are known to be prevalent, more than 90% of patients are aged less than 10 years old at the time of diagnosis[4]. pPNETs are commonly reported in the chest and paravertebral regions; however, atypical involvement, such as of retroperitoneal organs, the lungs, testes and uterus, is also seen[5].
According to the latest soft tissue and bone tumours classification, presented by the World Health Organization (WHO), PNETs are considered to be Ewing sarcomas, since identical chromosomal translocation (t(11;22)(q24;q12)) has been detected in Ewing sarcomas and in more than 85% of PNETs[4, 6]. Furthermore, PNETs located peripherally typically express high amounts of the MIC2 antigen (CD99) [7]. However, recent updates have suggested heterogeneity in retroperitoneal PNET presentation, which is a combination of a pPNET with non-differentiated retroperitoneal sarcomas [8]. Hence, the ambiguous and vague presentation of the disease contributes to a complicated diagnosis. As a result, appropriate treatment is a crucial challenge for these patients due to late referral. In the present report, we describe our experience with a 44-year-old male with abnormal retroperitoneal PNETs in our hospital, who was operated on with a spindle cell neoplasm diagnosis.
CASE DESCRIPTION
In May 2019, a 44-year-old man with acute severe pain in the right flank, which prohibited him from standing upright, presented to the emergency room. With respect to the illness manifestations, he had a complaint of mild pain in the same region from 2 weeks previously, accompanied by no other signs or symptoms. During the physical examination, severe right costovertebral angle (CVA) tenderness was detected. Primary laboratory testing during the first admission revealed a slightly elevated white blood cell count (10,110), mildly elevated C-reactive protein (CRP; 7 mg/l) levels and a normal erythrocyte sedimentation rate (ESR; 11 mm/hr).
Abdominopelvic ultrasonography examination showed a standard size, shape and parenchymal echogenicity for the liver, spleen and kidneys; however, a cortical cyst was detected on the upper pole of the left kidney with dimensions of 25×24 mm. Otherwise, no significant ultrasonographic findings were reported.
Spiral abdominopelvic computed tomography (CT) scans, performed with and without intravenous and oral contrast, showed a mildly calcified lesion with heterogenous density, 60 mm in length and 42 mm in diameter, on the projection of the inferior vena cava (IVC) at the level of the renal veins that continued to the IVC pathway, resulting in a caudal dilatation of the IVC. The CT scan findings were suggestive of aneurysmal dilatation of the IVC with internal thrombosis. Subsequently, the patient was subjected to magnetic resonance imaging (MRI) of the abdominal and pelvic cavities, which showed a lobulated-border, heterogeneously enhancing mass of 44×56×58 mm in size with a heterogeneous signal. MRI demonstrated enhancing septa and enhancing restricted peripheral-located soft tissue in an anatomical location between the psoas and vertebral body, and retrocaval, with invasion of the adjacent IVC at the infrarenal level (Fig. 1). The differential diagnoses were malignant pheochromocytoma, paraganglioma and retroperitoneal sarcoma.
The patient then underwent CT-guided core needle biopsy and immunohistochemical investigation of the retroperitoneal mass showed a predominance of stratified muscle fibres and fibrous connective tissue mixed with the separated fragments of a small cell tumour with focal necrosis.The tumour cells were positive for CD56 and FLI1 markers, as well as for CD99 (they were negative for LCA, CgA, Syn, desmin, myogenin, CD34 and TdT). Although complete evaluation was not possible due to insufficient tumoral tissue, the pathologist suggested taking PNET, as the highly probable diagnosis, into consideration (Fig. 2).
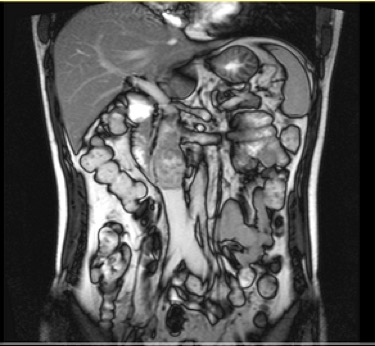
Figure 1 (click to enlarge)
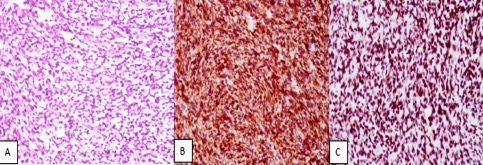
Figure 2 (click to enlarge)
Figure 1. Radiology: abdominal MRI, with IV and oral contrast, coronal view.
Figure 2. Pathology: (A) sheet-like proliferation of small round cells (H&E); (B) membranous reaction with CD99; (C) positive nuclear staining for FLI1
The insufficient sample required a repeat CT-guided core needle biopsy, which revealed neoplastic spindle cells by negative immunostaining for PanCK, CAM 5.2, DOG1, c-kit, CD34 and S100, while cells were equivocally positive for CD99 and desmin staining.The morphologic and immunohistochemistry findings were not supportive of the differential diagnoses of a gastrointestinal stromal tumour (GIST), the Ewing family of tumours (EFT) or sarcomatoid carcinoma. In addition, there are some morphologic overlaps between benign spindle cell proliferation and low-grade or locally invasive tumours such as fibromatosis, especially with a core needle biopsy specimen, that urged clinicians to follow the clinical and imaging findings for decision-making.
On July 2019, a total tumour resection was carried out via a laparotomy. The duodenum was rotated medially using a Kocher manoeuvre, and the tumour with the adjacent IVC section was explored. The overlying peritoneum was then dissected, and the tumour location was indicated; it originated from the IVC just below the branching of the renal veins and extended 4–5 cm towards the iliac vein bifurcation. Regarding the tumour invasion of the IVC, directly after separating the adhered tumour from the aorta, 2,500 IU of intravenous heparin was injected. The distal end of the IVC was resected with an appropriate margin 3 cm proximal to the iliac bifurcation. Subsequently, the tumour was sequestered from the posterior wall muscles and vertebral spine, which was followed by resection of the IVC proximally, inferior to the level of the renal veins, as well as the right renal artery, which was involved by the tumour. Accordingly, the left renal vein was damaged during IVC resection, which was repaired by 5-0 prolene. The IVC was replaced using an 8-8-16 Dacron graft via an end-to-end anastomosis.
Postoperative histopathological evaluation confirmed a PNET diagnosis. Patient recovery was uneventful and he was discharged without complications. Although the right kidney was non-functional, creatinine levels were within the normal range. Afterwards, the patient was referred to receive adjuvant chemotherapy with respect to the pathological outcomes.
DISCUSSION
PNETs belong to the Ewing sarcoma family of tumours (ESFTs), which was first described in 1921 by James Ewing from the case of a 14-year-old female with a malignant bone tumour composed of small round cells[9]. PNETs are considered to be highly malignant tumours composed of small round cells originating from neuroectodermal tissue that attack the central nervous system or peripheral organs, varying in clinical presentation based on the origin of the tumour[10]. Usually, radiological and pathological work-ups are associated with misdiagnosis[11]. In contrast to central PNETs that arise in the central nervous system, neural crest cells are responsible for the development of pPNETs[12]. Furthermore, standard treatment approaches do not exist for this disease, due to its rarity.
To the best of our knowledge, there are approximately 37 cases reported in the literature of patients who suffered from retroperitoneal PNETs. Thus, our awareness of appropriate diagnostic methods, approaches and efficient treatments is insufficient, and many studies and investigations are still required to recognize the different disease manifestations, signs and symptoms and features. In the current report, we describe a case of pPNETs in an adult who presented with sudden-onset severe abdominal pain and CVA tenderness. During the primary assessment, the patient had a retroperitoneal and retrocaval mass at the level of the renal vessels, which had ambiguous and indefinite pathological features that prohibited the pathologists and clinicians from acquiring an accurate diagnosis before the operation. A noteworthy feature of our patient’s case was the IVC involvement by the tumour, despite previous reports: IVC invasion has been reported to be a rare complication of retroperitoneal pPNETs[13].
Furthermore, during the imaging examination, the CT scan revealed a well-defined mass with heterogeneity that represented a mild calcification on some regions; however, no signs of irregularity, poor definition or hypodensity were observed, all of which have previously been reported as the most common findings[14]. Despite the fact that the CT scan appearance of PNETs is nonspecific for the lesion diagnosis, adjacent organ displacement and dislocation is also expected. Although the microcalcification could have developed due to necrotic tissue generation inside the lesion, these findings are not expected in patients suffering from retroperitoneal PNETs, and tissue necrosis has regularly resulted in heterogeneous enhancement of the lesion during contrast administration rather than being a classification sign. Nevertheless, the findings of the MRI tumour evaluation were in line with the literature, with PNETs usually presenting as an isointense to hypointense lesion on MRI [15].
In terms of pathologic assessment of the tumour, there was a remarkable difference in the outcomes of the histopathologic studies obtained from the first and second core needle biopsies. The first biopsy showed small round cells positive for CD99, CD56 and FLI1 cell surface markers, which was compatible with the histopathologic features of PNETs [16, 17]. In addition, other differential diagnoses were taken into consideration, such as rhabdomyosarcoma, neuroblastoma and non-Hodgkin lymphoma, but the primary consideration was in favour of a PNET. However, the second set of biopsy results completely supported the first differential diagnosis, and spindle cell sarcoma, with negative immunohistochemistry staining for nearly all tumour markers being the main finding of the evaluation. Therefore, the patient was indicated to undergo excisional surgery with the sarcoma considered to be the primary diagnosis. Ultimately, postoperative evaluation failed to prove the sarcoma diagnosis, and a PNET was confirmed by assessing the excised tumour. PNETs express histopathological characteristics that have an 85% similarity to Ewing sarcomas; however, the second biopsy rejected the diagnosis of a PNET. We failed to establish a correlation between the different pathological findings with respect to this unique tumour. Some hypotheses may rationalize these manifestations as follows: first, despite the low probability, it should be kept in mind that the patient may have been suffering simultaneously from a sarcoma and a retroperitoneal PNET; second, the current patient presented with an atypical type of PNET that developed a combination of PNET and sarcoma expression.
CONCLUSION
Despite the poor prognosis in patients suffering from PNETs, multimodal treatments have been suggested to improve survival. Considering recent reports emphasizing the role of neoadjuvant chemotherapy in the management of PNETs, patients underwent subsequent adjuvant chemotherapy after surgical resection [18–20]. In the short interval between surgery and the preparation of our report here, the patient in this case has had an uneventful recovery and no signs of recurrence have been noted.