ABSTRACT
Kikuchi-Fujimoto disease (KFD) is a rare, benign and usually self-limiting disorder that more often affects young women, which is characterized by cervical lymphadenopathy and fever. Clinical presentation may be indistinguishable from other diseases, and its inclusion in the differential diagnosis of lymphoproliferative, infective and autoimmune diseases is essential. An association with systemic lupus erythematosus is acknowledged. We present 2 different cases of 2 young women with KFD; the first case highlights the classic diagnostic features of this rare entity, and the second, the findings when KFD occurs in association with systemic lupus erythematosus.
LEARNING POINTS
- Fever and cervical lymphadenopathy presenting in a young woman are the main features of KFD.
- A timely excisional lymph node biopsy is of the utmost importance in establishing a correct diagnosis and in the management of this condition.
- In most patients with KFD the course of the disease is benign and self-limiting; however, when associated with other conditions (mainly systemic lupus erythematosus) it can follow a more severe evolution.
KEYWORDS
Kikuchi-Fujimoto disease, histiocytic necrotizing lymphadenitis, cervical lymphadenopathy, systemic lupus erythematosus, corticosteroids
CASE DESCRIPTION
Case 1
A 26-year-old Caucasian woman was referred to the Hematology Consultation due to cervical adenopathies of 1 month’s duration. She also complained of fever, night sweats, weight loss (10 kg in 3 months) and asthenia with no anorexia. Apart from a past medical history of breast fibroadenoma and recurrent tonsillitis (no episodes in the previous 4 months), she was otherwise healthy. On physical examination, she had multiple slightly tender, firm, bilateral cervical adenopathies, measuring approximately 2–3 cm; the remainder of the examination was unremarkable, with no hepatosplenomegaly. Investigation revealed minor leucopenia and thrombocytopenia, a slightly elevated erythrocyte sedimentation rate (ESR; 33 mm/h) and C-reactive protein (CRP) levels (1.15 mg/dl), normal lactate dehydrogenase (LDH), a normal autoimmune panel and negative serologies. Abdominal ultrasound was normal. 18F-fluorodeoxyglucose positron emission tomography/computed tomography (18F-FDG PET/CT) was perfomed and showed enlarged hypermetabolic supradiaphragmatic nodes with a maximum standardized uptake value of 7.6, indicating high-grade lymphomatous lesion activity. The final diagnosis was established based on histopathological results from the excisional biopsy of a lymph node that showed partial distortion of the architecture with extensive paracortical necrotic areas and abundant karyorrhectic debris, with proliferation of crescentic histiocytes and plasmacytoid monocytes, in the absence of polymorphonuclear neutrophils. Immunohistochemical studies – CD3+, CD5+, CD4+ and CD8+, predominance of CD8+, some CD20+ (peripheral), CD68+ and CD163+ (multiple histiocytoid cells) – corroborated the diagnosis of histiocytic necrotizing lymphadenitis (Kikuchi’s disease). A diagnosis of KFD was rendered and treatment with non-steroidal anti-inflammatory drugs (NSAIDs) for 2 weeks provided a favourable course, with complete resolution of clinical and laboratory signs. The patient is still undergoing active surveillance without evidence of recurrence.
Case 2
A 25-year-old Caucasian female nurse was referred to the Internal Medicine Consultation presenting with complaints of low fever, asthenia, weight loss and cervical lymphadenopathies over the previous 3 months. She also presented with a history of 12 months of arthralgias and joint stiffness upon waking, with major involvement of the knees, wrists, ankles and proximal and distal interphalangeal joints. As she noticed worsening over the previous 3 months, she was taking ibuprofen almost every morning. There was no other regular medication. There was a past medical history of allergic rhinitis and atopic dermatitis and a family medical history of type 1 diabetes mellitus, vitiligo and rheumatoid arthritis. On physical examination, there were several tender bilateral submandibular, supraclavicular and posterior cervical, right axillary and inguinal adenopathies, most of these being 2 cm in diameter, and she had pruritic erythematous cutaneous lesions on the anterior thoracic and posterior cervical areas and shoulders. Her previous laboratory tests revealed mild leucopenia, normocytic normochromic anaemia, an elevated ESR (68 mm/h) and CRP levels (0.82 mg/dl), low complement levels and antinuclear antibody (ANA) positivity. Additional laboratory investigation, bone marrow studies and an excisional biopsy of a cervical lymph node were conducted. However, the patient’s clinical status deteriorated and she was hospitalized. She looked unwell, with severe asthenia, pallor, but was anicteric. Laboratory assessment at that time showed severe haemolytic anaemia (Table 1). A thoraco-abdomino-pelvic computed tomography (CT) scan confirmed multiple adenopathies – axillary, mediastinal, coeliac, mesenteric and inguinal – that ranged in size from 1 to 2.5 cm and showed mild hepatomegaly and a small amount of fluid in the rectouterine pouch. In addition, serology tests for syphilis, human immunodeficiency virus (HIV), hepatitis B virus and hepatitis C virus, Epstein-Barr virus, cytomegalovirus, toxoplasmosis, borreliosis and QuantiFERON TB testing were negative. β2-microglobulin levels were increased, serum protein electrophoresis revealed a non-monoclonal increase in the gamma region and serum complement levels were greatly diminished. A positive immunological work-up corroborated the diagnosis of systemic lupus erythematosus (SLE) (em>Table 2). Bone marrow studies revealed normocellular marrow with adequate trilineage haematopoiesis with no Reed-Sternberg cells.
Due to the severity of the clinical condition with decreasing haemoglobin levels, imperative treatment with corticosteroid therapy at a high dose was started (methylprednisolone 1.5 mg/kg/day for 5 days), while we were waiting for the histological results.
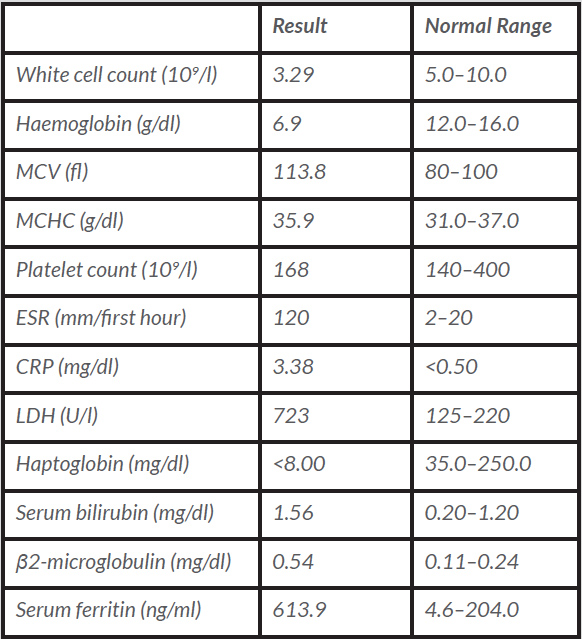
Table 1 (click to enlarge)
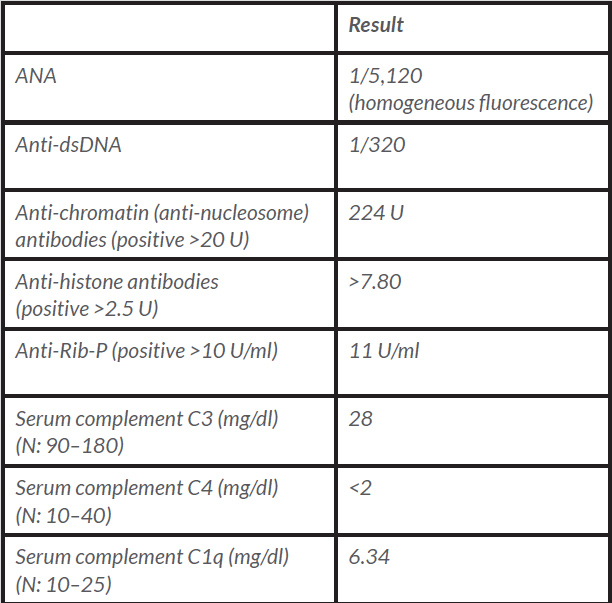
Table 2 (click to enlarge)
Table 1. The biochemical results in the deterioration phase of Patient 2
MCV: mean corpuscular volume; MCHC: mean corpuscular haemoglobin concentration; ESR: erythrocyte sedimentation rate; CRP: C-reactive protein; LDH: lactate dehydrogenase.
Table 2. Immunological results for Patient 2
ANA: antinuclear antibody; anti-dsDNA: anti-double-stranded deoxyribonucleic acid antibodies; anti-Rib-P: anti-ribosomal P protein antibodies; N: normal values.
Nevertheless, these results came back inconclusive and a second excisional biopsy of a cervical lymph node was performed (even though the patient had already initiated steroid therapy and so the results could have been difficult to interpret). Histological examination revealed cellular infiltrates composed of foamy histiocytes, with areas of coagulative necrosis, compatible with a diagnosis of histiocytic necrotizing lymphadenitis (Fig. 1), although at this point a diagnosis of SLE-associated lymphadenitis cannot be excluded. A skin biopsy was also performed but the results were inconclusive.
The patient’s symptoms markedly improved but she developed tachycardia and a newly emerged grade 3/6 systolic murmur over the precordial area and left sternal border. A 2-dimensional echocardiogram revealed: normal left ventricle size, normal left ventricle systolic function, concentric hypertrophy involving the interventricular septum (16 mm), the presence of systolic anterior motion (SAM) and clinically relevant left ventricular outflow tract obstruction (LVOTO) with a maximal systolic gradient of 50 mmHg with moderate mitral regurgitation and mild pericardial effusion. Due to the hypertrophic cardiomyopathy findings, beta-blocker therapy was initiated. After 6 months, the echocardiographic findings improved. In conclusion, a diagnosis of KFD associated with SLE was established. Overall, the patient presented a favourable clinical outcome with the given treatment – a tapering regimen of glucocorticoids, azathioprine and hydroxychloroquine (HCQ).
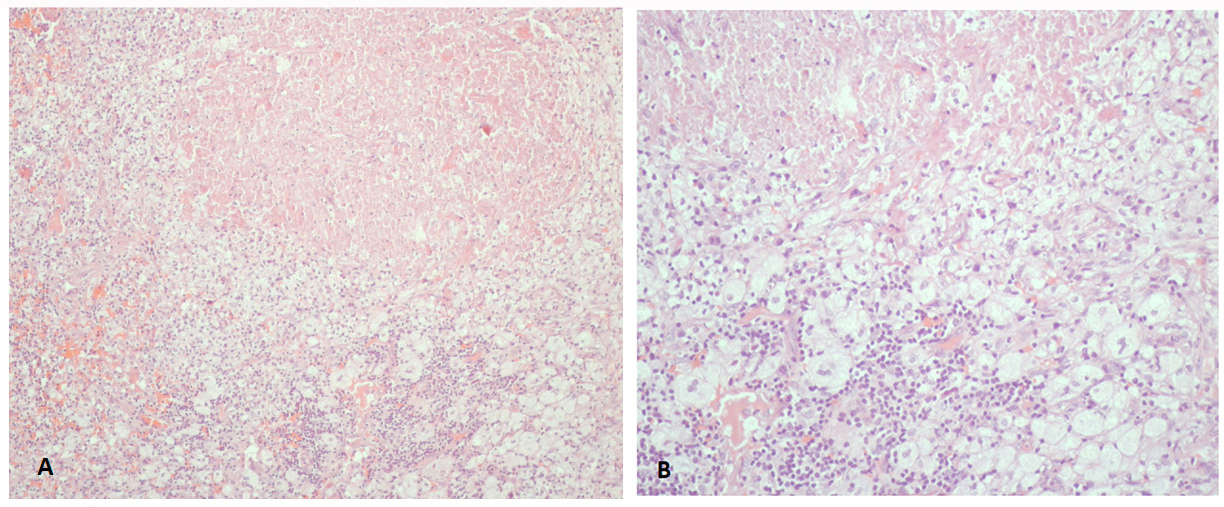
Figure 1 (click to enlarge)
Figure 1. Photographs of histological findings showing cellular infiltrates composed of foamy histiocytes, with areas of coagulative necrosis. (A) H&E staining x100; (B) x400
DISCUSSION
We report 2 clinical cases with the same diagnosis but with a very different clinical course.
KFD was first described in Japan almost simultaneously by Kikuchi and by Fujimoto and colleagues in 1972[1–5]. KFD is an extremely rare disease[1] and although it can affect both sexes and all age groups, it occurs more often in young women (<40 years of age).
The precise aetiology remains unknown, although several types of infection have been hypothesized to act as causal agents. Some authors have proposed local hyperimmune stimulation after infection, influenced by individual genetic predisposition or autoimmune mechanisms, as a possible pathogenic feature[2,3].
KFD frequently manifests as an acute or subacute illness, evolving over a period of 2 to 3 weeks[3]. Cervical lymphadenopathies (70–100%), usually unilateral and tender, and fever (30–50%) are the most common symptoms in KFD patients[1,4]. Less commonly, patients can present with weight loss, night sweats, asthenia, arthralgias, cutaneous manifestations or hepatosplenomegaly. Rarely, generalized lymphadenopathies can occur (5% of cases)[5].
Laboratory tests are frequently unremarkable; however, a mild leucopenia, anaemia and, more rarely, thrombocytopenia can be found, as well as elevated inflammatory markers (CRP and ESR)[4].
Therefore, its inclusion in the differential diagnosis of infectious, lymphoproliferative, neoplastic and autoimmune diseases is imperative, the most common of these being tuberculous lymphadenitis, malignant haematologic diseases (non-Hodgkin lymphoma and Hodgkin disease) and SLE[5]. An initial misdiagnosis as malignant lymphoma is accomplished in 30% of cases[4].
The association of KFD with other pathologies has been reported, mostly autoimmune, and more commonly with SLE. The clinical, laboratory and histopathological features of these 2 diseases commonly overlap, making differentiation impossible in some cases. Patients with KFD should have long-term follow-up due to their increased risk of SLE[2,5].
A definitive diagnosis of KFD is made by lymph node excisional biopsy. The histological findings consist of histiocytic infiltrate, areas of focal necrosis, karyorrhoexis, with a typical absence of neutrophils and eosinophils[3,5]. Three evolving histologic stages are identified in KFD: the proliferative, with lymphoid hyperplasia, variable histiocytic infiltrate and karyorrhexis; the necrotizing, when extensive paracortical or interfollicular necrosis is recognized and the xanthomatous, if there is a predominance of foamy histiocytes[3]. Immunohistochemical analysis constitutes an additional aid to diagnosis.
In most patients with KFD, the clinical course is benign and self-limiting. Symptomatic patients can benefit from NSAIDs for a short period of time or, in more severe cases, from corticosteroids, HCQ or intravenous immunoglobulin[3,4].
Our first clinical report highlights the most common features of a KFD patient: a polyadenopathic condition presenting in a young woman, adenopathies mainly localized to the cervical area, with a maximum diameter of 4 cm and tender, with constitutional symptoms. The laboratory findings were also consistent with what is reported in the literature. In most cases, rarely seen KFD is not the most likely diagnosis, but an accurate diagnosis is imperative, as this can prevent unwarranted and deleterious treatments; for example, if a misdiagnosis of a lymphoproliferative disease were to be made. A timely excisional lymph node biopsy was of the utmost importance in establishing a correct diagnosis and in the management of this condition.
In our second case, a diagnosis of KFD associated with SLE was established; thus, the patient did not have a straightforward clinical presentation. The Systemic Lupus International Collaborating Clinics (SLICC) criteria proposed in 2012 warranted the SLE diagnosis at that time as 4 clinical criteria (synovitis, serositis presenting as pericardial effusion, haemolytic anaemia and leucopenia) and 4 immunologic criteria (ANA+, anti-dsDNA+, antiphospholipid Ab and low complement) were present. The diagnosis remains using the new 2019 SLE criteria. Due to the evidence of haemolysis leading to the clinical deterioration of our patient, a more agressive pharmacological approach was imperative.
The second excisional biopsy of a cervical lymph node rendered the diagnosis. This condition is often undiagnosed or misdiagnosed as sometimes the histological characteristics of KFD are indistinguishable from those found with other entities, especially in the early stages of the disease
We also emphasize the hypertrophic cardiomyopathy findings in this young patient without a familial history of heart disease or sudden cardiac death. Jiang et al. reported a case of a 25-year-old woman with SLE with myocardial hypertrophy that was assumed to be related to high-dose prednisolone use[6]. Perhaps this could be the explanation for our patient’s unexpected clinical finding.
In conclusion, with this report we would like to increase medical awareness of this under-recognized entity, as it is a crucial inclusion in the differential diagnosis in cases of lymphadenopathy and fever.