ABSTRACT
Introduction: Kikuchi-Fujimoto (KF) disease is a rare and self-limiting disorder of unknown aetiology characterized by cervical lymphadenopathy (CLN) and fever. The pathophysiology remains unclear and may be triggered by an infectious agent leading to a self-limiting autoimmune process. There are no confirmatory laboratory tests and lymph node biopsy is required to differentiate KF disease from other serious conditions.
Materials and methods: We report 11 cases of KF disease diagnosed at Hamad General Hospital, Qatar, between 2006 to 2016. The diagnosis is based on clinical presentation, investigations and histopathological examination of lymph nodes.
Results: All patients had painful neck swelling (average duration of 2.9 weeks) and 10 had fever (average duration of 3.2 weeks). Five patients developed mild leucopenia which resolved completely. HIV and tuberculosis (TB) screening including sputum for AFB, a PPD skin test and chest x-ray was done for all patients and came back negative. Autoimmune screening was done for all patients and excluded any rheumatological disease. Ultrasound and CT of the neck confirmed cervical lymphadenopathy. Except for hepatomegaly in one patient, CT scans of the chest and abdomen were negative for any lymphadenopathy or organomegaly (performed in seven patients). Diagnosis was confirmed on lymph node excision biopsy. Histopathological examination showed findings consistent with the diagnosis of histiocytic necrotizing lymphadenitis (KF disease).
Conclusion: KF disease should be kept in mind for patients presenting with fever and CLN. Lymphoma, TB and autoimmune diseases like systemic lupus erythematosus should be excluded in such patients.
LEARNING POINTS
- Kikuchi-Fujimoto (KF) disease should be kept in mind in patients presenting with fever and cervical lymphadenopathy with or without other constitutional symptoms.
- There are no confirmatory blood tests and KF disease requires histopathological examination of involved lymph nodes for confirmation.
- Recurrence of disease, although rare, is a possibility and needs histopathology for confirmation.
- Recurrence of disease, although rare, is a possibility and needs histopathology for confirmation.
KEYWORDS
Kikuchi-Fujimoto disease, histiocytic necrotizing lymphadenitis, lymphadenopathy
INTRODUCTION
Kikuchi-Fujimoto (KF) disease, also known as histiocytic necrotizing lymphadenitis, is a rare, benign, and self-limiting disease of unknown aetiology. This condition was first described by Kikuchi and Fujimoto in Japan in 1972[1]. It is more prevalent among young Asian females, although cases are reported from Europe and the American region as well. It mainly effects young adults but a few paediatric cases have been reported [2, 3]. The most common presentation of KF disease is painful lymphadenopathy (mainly cervical), fever and upper respiratory tract symptoms. Other less common symptoms are skin rash, headache, night sweats, weight loss and arthralgia[4]. The aetiology remains unclear, but it is probably triggered by a virus leading to a self-limiting autoimmune process[5]. There is no confirmatory laboratory test. High levels of C-reactive protein (CRP) and ferritin, mild cytopenia and raised lactic dehydrogenase (LDH) are often noted. Although KF disease is frequently attributed to the autoimmune process, serological tests for auto-antibodies are usually negative[6]. Diagnosis of KF disease is confirmed by biopsy of involved lymph nodes which typically shows necrosis, karyorrhexis and histiocyte accumulation at the edge of necrosis[7]. Prognosis is excellent and most patients recover with supportive care[8].
In this report, we describe an illustrative case with recurrent KF disease and present a collective series of 11 cases of KF disease diagnosed in the Department of Medicine of Hamad General Hospital, Qatar.
CASE DESCRIPTION
Lymph node biopsy specimens diagnosed as histiocytic necrotizing lymphadenitis (KF disease) by the Histopathology Department at Hamad General Hospital between 2006 and 2016, were retrieved. These were from 11 patients who presented with lymphadenopathy mainly involving the cervical area and fever. Clinical data including age, gender, nationality, clinical presentation, investigations and treatment were recorded. We describe an illustrative case with recurrent disease in detail and present the collective data from all 11 cases in Table 1.
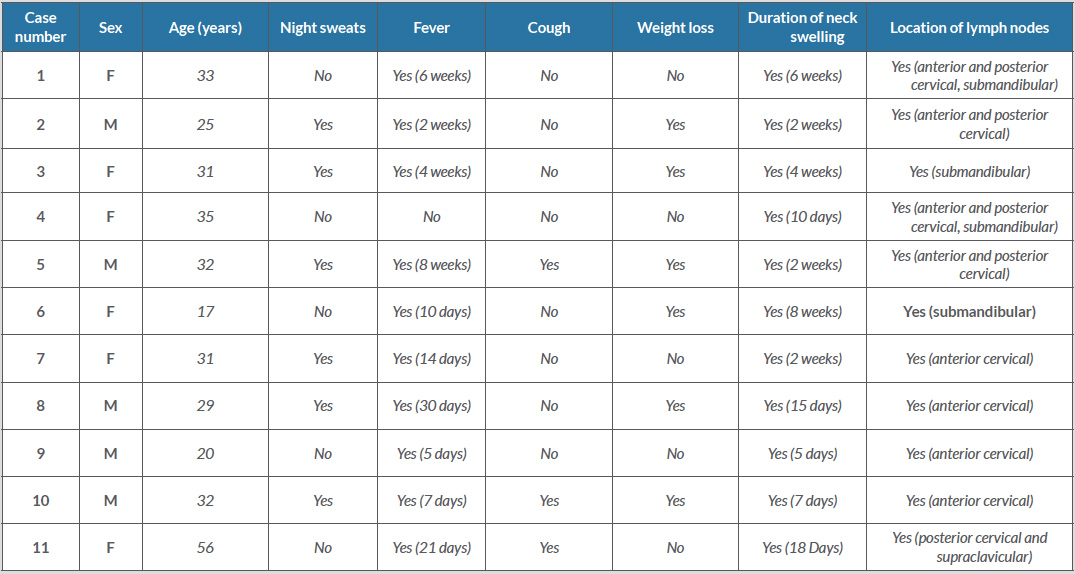
Table 1 (click to enlarge)
Table 1. Symptoms and physical findings
Illustrative Case with Recurrent Disease
A 31-year old woman, with a medical history of KF disease 9 years previously, presented with fever for 14 days and skin rash for 2 days. She had gestational amenorrhoea of 18 weeks at the time of presentation. There was no history of cough, weight loss, night sweats, nausea, vomiting, abdominal pain, joint pain, joint stiffness or swelling in any part of the body. Physical examination showed tender right-sided cervical lymphadenopathy, a morbilliform rash on the limbs and some scattered rashes on the trunk. Abdominal examination did not reveal any visceral enlargement. Heart, chest and nervous system examinations were normal. Laboratory investigations showed haemoglobin of 12.9 (13–17 g/dl), WBC count 4.6 (4–10), platelet count 253 (150-400), urea 2.3 (2.50–6.40 mmol/l), creatinine 42 (64–110 µmol/l), bilirubin 8 (up to 24 µmol/l), alkaline phosphatase 56 (45–129 U/l), ALT 64 (0–30 U/l) and AST 24 (0–31). The serum LDH level was 195 (125–220 U/l) and CRP was 15 (0–5 mg/l). A peripheral smear did not show any abnormalities. Blood cultures, urine culture, ANA profile, and screening for HIV, HBV and HCV were all negative. IgM for rubella, varicella, EBV, measles and parvovirus were also negative. Recurrence of KF disease was confirmed on excisional biopsy of a cervical lymph node which showed foci of partial coagulative necrosis with abundant karyorrhexic debris containing a variety of cells including histiocytes, plasmacytoid monocytes, some immunoblasts and some small and large lymphocytes. There were also plasma cells. There were no neutrophils and no definite granulomata. The patient recovered completely and had an uneventful pregnancy outcome.
All Cases
There were five males and six females in our series. These included three Qatari, two Indian, two Bangladeshi, one Nepalese, one Indonesian, one Mauritanian and one Moroccan patient. None of the patients were related to each other. All the non-Qatari patients were expatriates working in different capacities in Qatar. Most patients were young with a median age of 31 years (range 17–56) years. All patients had painful neck swelling (average duration of 2.9 weeks) and 10 had fever (average duration of 3.2 weeks). Table 1 shows the demographic characteristics and clinical features at presentation of all 11 cases.
Five patients developed mild leucopenia which resolved completely. Details of full blood counts and inflammatory markers are shown in Table 2. HIV and TB screening including sputum for AFB, a PPD skin test and chest x-ray was done for all patients and came back negative. Autoimmune screening was done for all patients and results are shown in Table 3.
Ultrasound and CT of the neck confirmed cervical lymphadenopathy. Except for hepatomegaly in one patient, CT scans of the chest and abdomen were negative for any lymphadenopathy or organomegaly (performed in seven patients). Diagnosis was confirmed on lymph node excision biopsy (Fig. 1). Histopathological examination showed findings consistent with a diagnosis of KF disease.
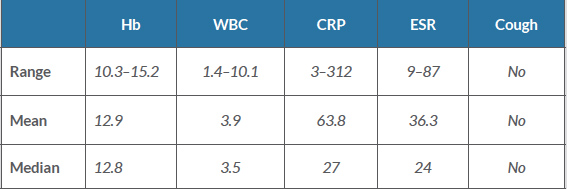
Table 2 (click to enlarge)

Table 3 (click to enlarge)
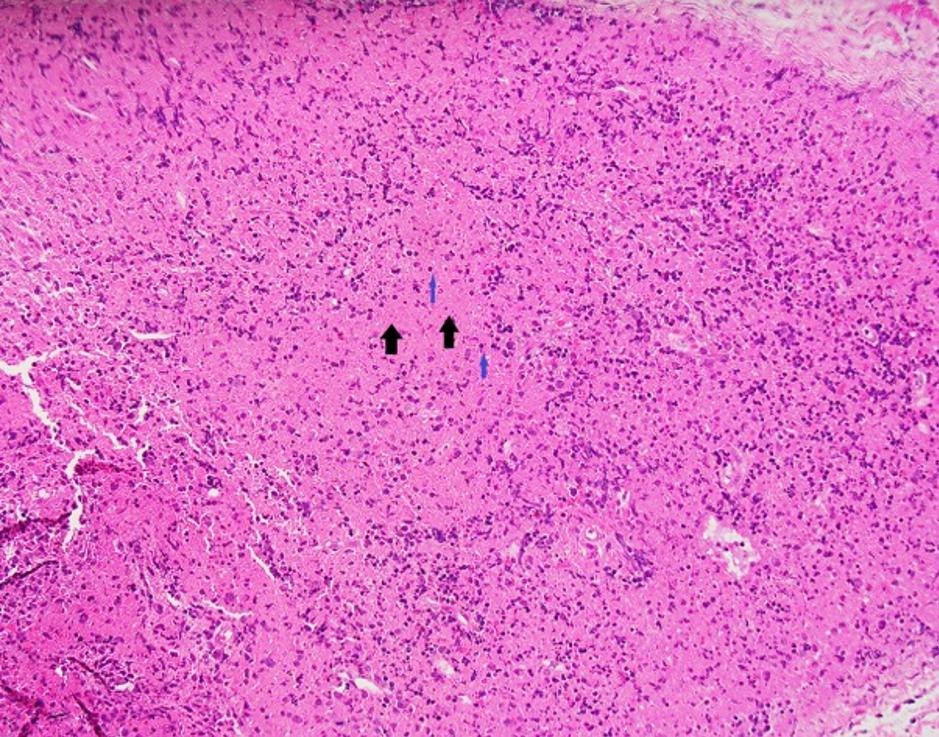
Figure 1 (click to enlarge)
Table 2. Laboratory results
CRP: C-reactive protein; ESR: erythrocyte sedimentation rate; Hb: haemoglobin; WBC: white blood cells.
Table 3. Autoimmune screen results
ANA: antinuclear antigen; ds-DNA: double-stranded deoxyribonucleic acid; ENA: extractable nuclear antigen; RA factor: rheumatoid factor
Figure 1. Section showing a lymph node with areas of necrosis (black arrows) and abundant karyorrhexic debris (blue arrows) containing a variety of cells including histiocytes and some small and large lymphocytes. There are no neutrophils and no definite granulomata. Findings are suggestive of histiocytic necrotizing lymphadenitis (Kikuchi–Fujimoto’s disease)
DISCUSSION
KF disease typically effects young adults. The median age of our patients was 31 years, which is consistent with other reported case series[9]. KF disease is more common in Asian females[9]. The number of male and female patients in our series is likely explained by the male predominance (75% of total) of the Qatari population.
All our patients presented with cervical lymphadenopathy and fever, which is the most common presentation of KF disease. The differential diagnosis of these symptoms is extensive, and includes TB, acute viral illnesses like infectious mononucleosis and AIDS, lymphoma, metastatic cancer and autoimmune disease. A hyper-immune response of T-lymphocytes to an unidentified agent is thought to be the underlying cause of KF disease[5]. Patients are screened for different infectious agents but no sole cause has been identified so far. Screening for TB (sputum for AFB, PPD testing and chest x-ray), HIV, EBV and CMV was negative in all of our cases as well.
Mild cytopenia and raised inflammatory markers are reported in patients with KF disease[9]. Five of our patients developed mild leucopenia that resolved completely. Review of literature suggests that generalized lymphadenopathy is very rare[10]. CT scans of neck, chest and abdomen done in seven of our patients showed lymphadenopathy in the neck area only. One patient showed hepatomegaly.
An association with autoimmune disease, specifically systemic lupus erythematosus (SLE), has been documented. The clinical features of both diseases have similarities but serological tests for SLE showed inconsistent results. Antinuclear antibodies (ANA) were negative in eight of our patients. One patient had weakly positive ANA, while one had strongly positive ANA. Ds-DNA antibodies, rheumatoid factor and extractable nuclear antigen (ENA) were negative in all of our patients. These results are consistent with other reported cases of KF disease.
Diagnosis of KF disease is confirmed by histological examination of involved lymph nodes[9, 10]. KF disease has distinct features on histopathology but is sometimes mistaken for malignant lymphoma or SLE. All of our patients had KF disease confirmed on excision biopsy of lymph nodes.
Recurrence is reported to be 3–4% and death is extremely rare[8]. The majority of patients as reported in different case series recover with supportive care but a few need steroids and immune suppressants. Erfanian Taghvaei et al. reported a case of KF disease recurrence in a 32-year-old female patient. The first episode resolved with anti-inflammatory medication, while a second episode was treated with oral steroids for 3 months with complete resolution of symptoms and lymphadenopathy [11]. Blewitt et al. also reported a case with recurrence of KF disease 12 years after the initial episode. The patient was managed conservatively without any medication and complete recovery was achieved[12]. Two of our patients had recurrence, including one who had a recurrence during pregnancy (the illustrative case). She recovered well and had an uneventful pregnancy. All of our patients fully recovered with symptomatic supportive care.
In conclusion, KF disease should be considered in patients presenting with fever and lymphadenopathy, particularly cervical lymph node involvement. TB, lymphoma and autoimmune diseases need to be excluded in all cases. There is no diagnostic laboratory test and lymph node biopsy is needed to confirm the diagnosis. In most cases, KF disease has a benign course and requires only supportive care.