ABSTRACT
We describe a 62-year-old patient with a 4-year history of myelodysplasia who later developedstriking features that included massive splenomegaly, rapidly evolving visual loss and a sensorimotor polyneuropathy. This led us to consider the diagnosisof haemophagocytic lymphohistiocytosis (HLH). Upon further investigation, we found that he fulfilled the necessarydiagnostic criteria for HLH, including the presence of haemophagocytosis of erythroid precursors on bone marrow smear.
LEARNING POINTS
- Haemophagocytic lymphohistiocytosis (HLH) can be a rare complication of myelodysplastic syndromes.
- HLH is ultimately fatal if timely allogeneic stem cell transplantation is not performed.
- This case highlights the importance of considering the diagnosis of HLH in patients with myelodysplasia who present with unusual features.
KEYWORDS
Myelodysplastic syndromes, lymphohistiocytosis, hemophagocytic, polyneuropathy
INTRODUCTION
Haemophagocytic lymphohistiocytosis (HLH) is defined by the association of several criteria including fever, splenomegaly, bicytopenia or pancytopenia, hypofibrinogenaemia hypertriglyceridaemia and haemophagocytosis on examination of bone marrow or spleen. Additional criteria include low naturalkiller (NK)-cell activity, hyperferritinaemia and high soluble interleukin-2-receptor levels[1].
HLH can be fatal if allogeneic stem cell transplantation (SCT) is not performed. Mortalityrates of 52% have been reported in secondary forms of HLH. Available treatment options include cytotoxic agents, corticosteroids, immunosuppressive therapyand SCT[2].
HLH may be idiopathic or it may be associated with several entities characterised byimmune dysregulation. Secondary forms of HLH may occur in the context of chronic infections and malignancy. HLH has beenstrongly associated with myelodysplasia[3].
We report the case of a 62-year-old patient with myelodysplasia who developed unusualfeatures and eventually fulfilled the criteria for HLH.
CASE REPORT
A 62-year-old patient had a 4-year history of refractory cytopenia with multilineal dysplasia. He also had a mild monoclonal IgG-lambda gammopathy. He was being treated with erythropoietin and G-CSF analogues. One year earlierhe had developed papilloedema and impaired vision. He improved with prednisone 60 mg/day and progressive tapering but his visiondeteriorated one month prior to admission and he had to be placed on the initial dose again.
He presented with a 4-day history of pain and swelling in his left leg. Deep vein thrombosiswas diagnosed through ultrasonography. He was placed on anticoagulation with dalteparin and the growth factors were temporarily withheld.
A CT scan of the chest and abdomen was performed to rule out an occult neoplasm. Itdemonstrated a splenomegaly of 17.4 cm that had progressively increased over the last year.
Treatment with growth factors was resumed 72 h after admission and we initially obtainedan adequate response. However, over the course of a month we observed progressive worsening of his pancytopenia.
A bone marrow aspirate demonstrated a hypercellular marrow with significant dyspoiesis in the erythroid and megakaryocytic series. While this smear ruled out progression to acuteleukaemia, it showed haemophagocytosis of erythroid precursors (Fig.1).
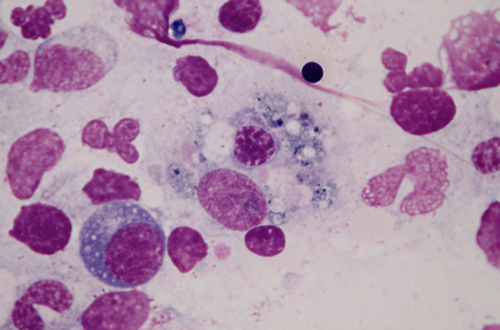
Figure 1 (click to enlarge)
Fig. 1 - Bone marrow aspirate showing phagocytosis of an erythroblast by a macrophage (x100, Giemsa stain).
Due to his worsening thrombocytopenia, with a platelet count that reached 5000/mm3, we withdrew treatment with dalteparin. Two weeks later he began to complain of pain and swelling in his left calf. Ultrasonography confirmed a newdeep vein thrombosis. We resumed anticoagulation.
The patient developed recurrent respiratory infections. A chest X-ray revealed a cavitating lesion in the right lower lobe. Pseudomonas aeruginosa was isolated from sputum and he was placed on meropenem and tobramycin (the patient had received tobramycin for 3 days at a dosage of 200 mg/twice daily). Aspergillus fumigatus was later isolated from sputum. He responded well to voriconazole and caspofungin.
Throughout his hospitalisation, his visual acuity deteriorated despite treatment. Markedmuscle wasting and progressive deafness also ensued. Orbital MRI ruled out leukaemic infiltration of optic nerves and nerve conduction studies revealed a sensorimotor polyneuropathy. Evoked potential testing wasconsistent with bilateral sensorineural hearing loss.
The patient progressively worsened and he reached a platelet count of 7000/mm3. Anticoagulant therapy was withdrawn due to bleeding risk. The patient died twomonths after admission. No autopsy was performed (Table 1).
| Variable | Reference range, adults | 3rd hospital day | 45th hospital day |
|---|---|---|---|
| Haemoglobin (g/dl) | 13.5-17.5 | 9.7 | 8.6 |
| Leukocytes (/mm3) | 4,500-11,100 | 7,800 | 9,000 |
| Neutrophils (%) | 50-60 | 83.9 | 96.1 |
| Lymphocytes (%) | 25-45 | 7.7 | 2.2 |
| Monocytes (%) | 3-10 | 8.3 | 1.7 |
| Basophils (%) | 0-1 | 0.1 | 0 |
| Eosinophils (%) | 1-4 | 0 | 0 |
| Platelets (/mm3) | 150,000-400,000 | 62,000 | 14,000 |
| Fibrinogen (mg/dl) | 220-450 | 423 | 153 |
| Triglycerides (mg/dl) | 50-200 | 230 | 308 |
| Ferritin (ng/ml) | 20-300 | 168 | 348 |
| Aspartate aminotransferase (U/l) | 0-40 | 14 | 16 |
| Alanine aminotransferase (U/l) | 0-40 | 16 | 30 |
| Alkaline phosphatase (U/l) | 40-129 | 76 | 160 |
| Gamma-glutamyl transpeptidase (U/l) | 7-40 | 34 | 71 |
Table 1 - Laboratory data
DISCUSSION
We describe a patient with severe myelodysplasia who had several remarkable features. He hada rapidly evolving splenomegaly, progressive visual loss and a sensorimotor polyneuropathy. These unusual features prompted us to consider the diagnosis ofan associated HLH. This scenario has rarely been described3. The diagnosis of HLH is based on several criteria[1], many of which were observedin this patient. Specifically, he had fever, pancytopenia, massive splenomegaly, hypofibrinogenaemia and hypertriglyceridaemia. We also found haemophagocytosis of erythroid precursors on bone marrow smear. Therefore, he fulfilled the five criteria required for diagnosis. Anotherdiagnostic criteria includes ferritin levels >500 ng/ml. However, the patient had hyperferritinaemia that did not reach this diagnosticthreshold.
Splenomegaly is very uncommon in myelodysplasia, and its presence may indicate myelomonocytic leukaemia, which was ruled out by bone marrow aspirate. Splenomegaly may constitute the first sign of leukaemic transformation. It may also indicate extensive extramedullar erythropoiesis[4]. Given the rarity of splenomegaly in myelodysplasia, this clinical feature would fit better withHLH.
Polyneuropathy has been more frequently reported in HLH than in myelodysplasia[5]. The papilloedema in this patient was perhaps the first sign of optic neuritis. The progression of optic nerve involvement was haltedby prednisone therapy, but later flared up when prednisone was tapered. MRI studies ruled out the presence of vascular lesions and leukaemic infiltration of optic nerve sheets.
Visual impairment has rarely been described in myelodysplasia; it may be causedby anaemic hypoxia and microvascular insufficiency. These mechanisms were probably not responsible for visual loss in this patientsince the optic neuritis was initially responsive to prednisone. Despite the presence of a mild monoclonal IgG-lambda gammopathy, there were no signs of a hyperviscosity syndrome.
Another striking feature of this patient was the development of progressivedeafness. A slight hearing impairment existed before admission, which worsened after being placed on tobramycin. Tobramycin was stopped after three days oftreatment but hearing loss continued to progress significantly.
Cranial neuropathies have been described in HLH. A retrospective study ofpatients with HLH revealed that as many as 73% had evidence of central nervous system (CNS) involvement at the time of diagnosis[5].
In summary, this patient developed clinical features compatible with HLH in thelast year of his life. When the diagnosis of HLH was considered, he had already developed lung aspergillosis and a severe polyneuropathy. This highlights the importance of considering the diagnosis ofHLH in patients with myelodysplasia and unusual features.