ABSTRACT
Pericardial effusion represents a diagnostic challenge. Erdheim-Chester disease (ECD), though a rare cause, should be considered in the differential diagnosis. An 88-year-old woman was admitted to the hospital due to retrosternal pain, dyspnoea and constitutional symptoms. Hypoxaemic respiratory failure and increased inflammatory markers were documented. A chest x-ray revealed an increased cardiothoracic ratio. An echocardiogram showed a moderate-volume pericardial effusion, without signs of cardiac tamponade. A thoraco-abdomino-pelvic CT scan found a bilateral perirenal soft tissue halo. Perirenal mass biopsy showed diffuse infiltration by foamy histiocytes (CD68+), without IgG4, compatible with ECD. The correlation of anamnesis, radiology and histology is crucial for the diagnosis of ECD.
LEARNING POINTS
- Erdheim-Chester disease is a non-Langerhans cell histiocytosis that affects multiple organs and systems.
- Thorough study of a pericardial effusion is important as it is still considered idiopathic in 10–20% of cases.
- It is a rare disease so high diagnostic suspicion is important. The diagnosis is established through clinical manifestations, radiologic findings and histological confirmation.
KEYWORDS
Erdheim-Chester disease, pericardial effusion, retroperitoneal space
CASE DESCRIPTION
An 88-year-old Caucasian woman with a history of ischaemic heart disease, New York Heart Association class II heart failure, paroxysmal atrial fibrillation with a pacemaker due to sinus node disease and essential thrombocytosis (with a JAK2 heterozygotic mutation), chronically medicated with hydroxyurea, acetylsalicylic acid and bisoprolol, was referred to the emergency department because of chest pain and dyspnoea.
It was an atypical retrosternal dull chest pain appearing mostly at night, self-limited, lasting 5 minutes, in the supine position, associated with paroxysmal nocturnal dyspnoea and worsening of the usual pattern of dyspnoea during the day. The patient also had weight loss (10% in 2 months), anorexia, asthenia and nausea ongoing for 2 months. She had no relevant epidemiological context nor family history.
Objectively, we noted mucocutaneous pallor, a normal arterial pressure profile, the jugular venous pulse was elevated to 12 cm of water above the right atrium, without Kussmaul’s sign. Arrhythmic and hypophonetic sounds were found on cardiac auscultation. Pulmonary examination revealed minimal crackles in the left base. The abdomen was soft and non-tender without hepatosplenomegaly. No cervical, inguinal or axillary lymphadenopathy was identified. There were no cutaneous nor neurological abnormal findings.
Methods and Procedures
Laboratory studies revealed increased inflammatory markers (leucocyte count of 24,800/μl with 80% neutrophils and C-reactive protein levels of 65 mg/l) and a platelet level of 859,000/ml. Blood gas analysis showed hypoxaemic respiratory failure (O2 partial pressure of 56 mmHg). An electrocardiogram showed atrial fibrillation without other remarkable findings and testing for myocardial necrosis markers was negative. A chest radiograph exhibited an increased cardiothoracic ratio. An echocardiogram demonstrated evidence of moderate-volume pericardial effusion, without signs of cardiac tamponade. At this point, the pericardial effusion was considered the most discriminatory finding and the most probable causes considered were neoplastic disease, namely leukaemic transformation of essential thrombocytosis, infectious disease, namely tuberculosis or an autoimmune disease, such as systemic lupus erythematosus. Nevertheless, the posterior location of the pericardial effusion made it inaccessible for pericardiocentesis. The peripheral blood smear had no immature cells, the sedimentation rate was 9 mm/1st hour while testing for antinuclear antibodies, antineutrophil cytoplasmic antibodies and HIV was negative. Endoscopic studies (both upper and lower) and mammography yielded no relevant findings. A thoraco-abdomino-pelvic CT scan showed a “coated aorta” (Fig. 1) and a bilateral perirenal soft tissue halo, with extension to the renal pelvis causing bilateral hydronephrosis (Fig. 2), and verified the presence of the pericardial effusion, mainly in a posterior location, confirming the risk of performing a pericardiocentesis.
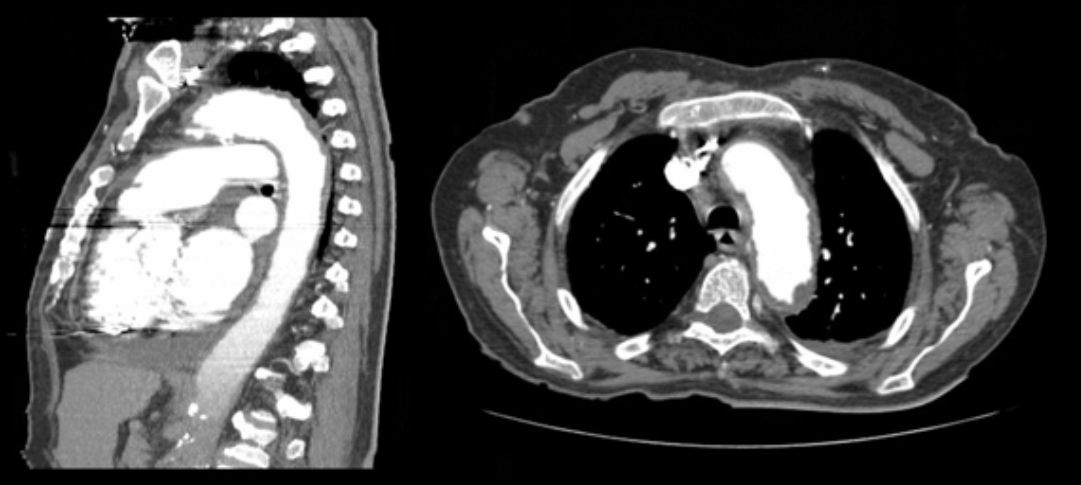
Figure 1 (click to enlarge)
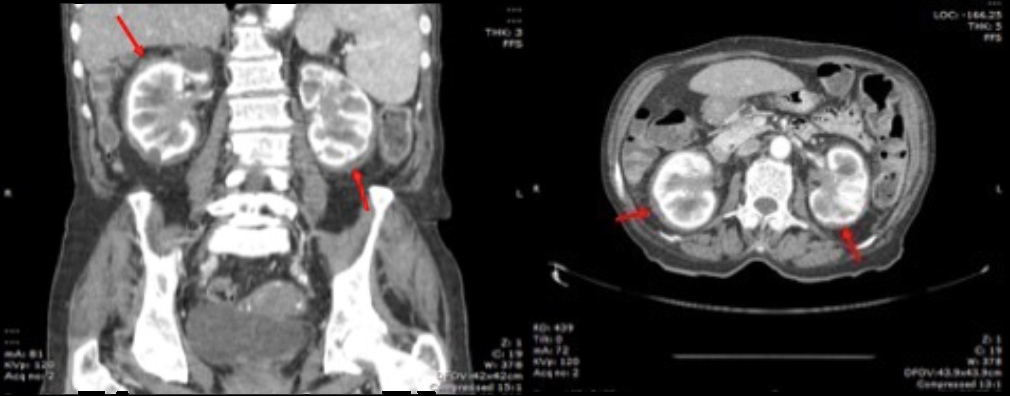
Figure 2 (click to enlarge)
Figure 1. Coated aorta (soft tissue halo around the aorta)
Figure 2. CT scan showing a soft tissue halo around both kidneys (red arrows) and bilateral hydronephrosis. The kidneys had a normal size and differentiation
These radiological images narrowed the differential diagnosis to renal lymphoma, IgG4-related disease and Erdheim-Chester disease (ECD). IgG quantification did not show a predominance of 1 serologic subclass. Bone scintigraphy showed discrete hyperactivity along the diaphysis of both femurs relatively symmetrical to each other, that could eventually translate some form of histiocytosis (Fig. 3). Perirenal mass biopsy was performed showing diffuse infiltration by foamy cytoplasm macrophages (CD68 positive) (Fig. 4), compatible with ECD. There was no evidence of lymphoma nor IgG4 deposition. Genetic study of the DNA of the mass cells did not find the p.Val600Glu pathogenic variant in the BRAF gene.
The patient was discharged as asymptomatic while waiting for definitive histology results. During the course of the outpatient evaluation, the patient presented functional decline and was readmitted with a urinary sepsis with no response to therapy, leading to the patient’s death.
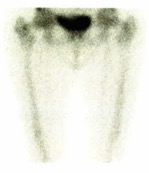
Figure 3 (click to enlarge)
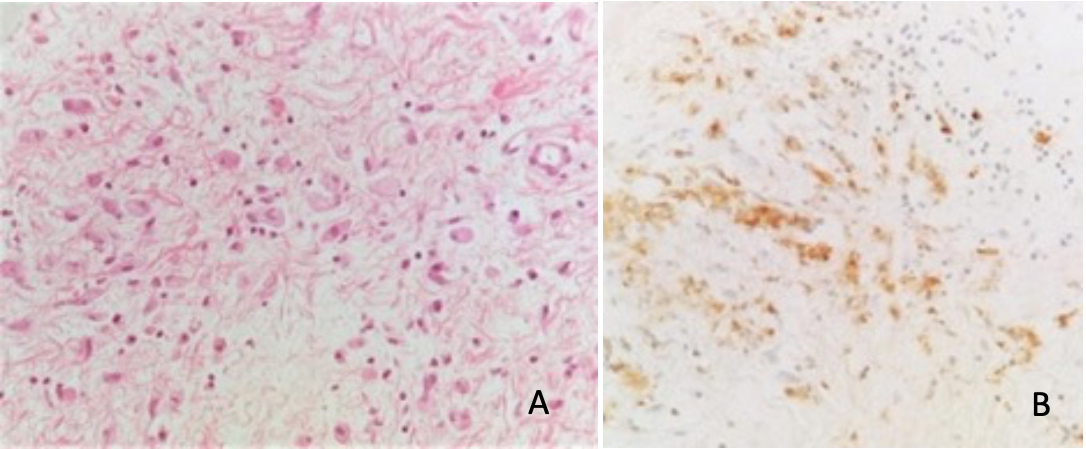
Figure 4 (click to enlarge)
Figure 3. Bone scintigraphy – bilateral femoral hyperactivity
Figure 4. (A) Dispersed multinucleated histiocytes with foamy cytoplasm. (B) Foamy histiocytes with CD68 immunoreactivity
DISCUSSION
ECD is a rare, non-Langerhans cell histiocytosis, characterized by systemic proliferation of and infiltration by foamy mononuclear phagocytic cells (histiocytes) [1–4]. It was first described in 1930 by Jakob Erdheim and William Chester [2, 4], and since then, more and more cases have been described, with some authors referring to 1,000 cases so far in the world. The disease has an onset between 40 and 70 years of age with a predominance in males [1, 2, 4].
It is a multisystemic disease that affects many parts of the body, including the skeleton, organs located in the retroperitoneum and the orbit, in addition to the cardiovascular, pulmonary, neurologic and endocrine systems [1–5], though the presentation may vary from asymptomatic to indolent focal disease to life-threatening organ failure [2, 4]. Some of the clinical manifestations are summarized in Fig. 5. Cardiovascular manifestations appear in 75% of patients [5] and include circumferential periaortic encasement, valvular disease, pseudotumoural infiltration of the chambers of the right side of the heart [1, 2] and pericardial disease. Involvement of the pericardium is the most frequent cardiac manifestation [5], occurring in 40 to 45% of patients, and can present with pericarditis or pericardial effusion with or without tamponade [2]. Retroperitoneal manifestations include infiltration of the aorta, causing periaortitis (“coated aorta”) [1, 5], and also of the perinephric tissues, leading to “hairy kidney”, ureteral narrowing and hydronephrosis [2, 4].
In this case, the most prominent clinical finding was a pericardial effusion; this always poses a challenging diagnostic path, as it demands the immediate and continuous evaluation of the haemodynamic consequences, as well as the search for a cause, which is even more difficult when the pericardiocentesis performance risk is high. The presence of unintentional clinically significant weight loss made it mandatory to look for a neoplastic or systemic autoimmune disease. In this patient, the main concern was the presence of a haematologic malignant disease, as these are the most commonly related to pericardial effusions and given the history of essential thrombocytosis in this patient, which has a risk of leukaemic transformation.
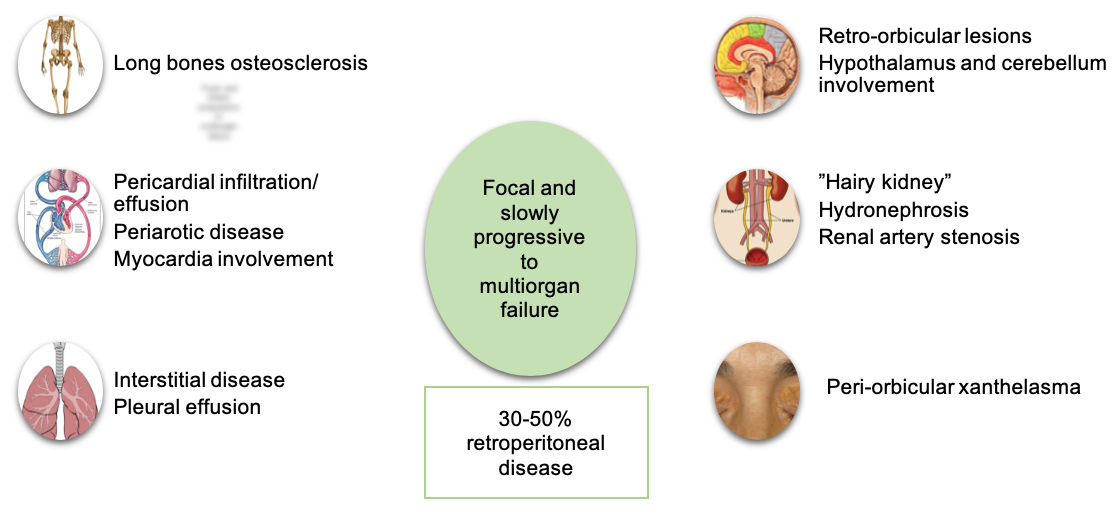
Figure 5 (click to enlarge)
Figure 5. Clinical manifestations of Erdheim-Chester disease
The hypothesis of an autoimmune disease was less likely due to the lack of other clinical findings, the sex and age of the patient and the absence of serologic markers. The search for a neoplastic disease began with the peripheral blood smear, followed by the endoscopic examinations, but only the realization of the CT scan was, in this case, sufficiently informative to narrow and guide the differential diagnosis, as it raised suspicion for less common entities in clinical practice.
Usually, imaging is helpful in establishing the diagnosis [1, 4], while laboratory findings are non-specific and non-diagnostic, typically including an elevated erythrocyte sedimentation rate and elevated C-reactive protein levels [4]. Common CT findings include the “coated aorta” and “hairy kidneys” [4], corresponding to histiocytic infiltration into the perirenal fascia [1], which is present in 68% of cases [2].
As in this case, the biopsy is usually the confirmatory examination, showing infiltrating CD68+, CD1a- foamy histiocytes [1, 2, 4, 6]. The BRAF V600E mutation was identified in many patients (prevalence ranging from 38 to 100%, according to the technique used [2, 4]), suggesting this disease may be a clonal disorder [2, 4] leading to the hyperactivation of proinflammatory cytokines [2], with a predominant Th1 phenotype [6], though the pathogenesis of the disease is still poorly understood. The association with this mutation motivated the treatment of these patients with vemurafenib, a BRAF V600E kinase inhibitor, which resulted in rapid and sustained regression of disease in some patients [1].
Although the aetiology is unknown, there is another case of ECD appearing after a myeloproliferative disorder [7], in that case polycythaemia vera, also with perinephric infiltration. The authors suggested that JAK2 somatic mutation may lead to a predisposition to acquire additional somatic mutations, including those of the BRAF gene, the activation of which is the final downstream component of the JAK-STAT pathway. They also postulated that the cytokine activation described in the myeloproliferative disease is similar to that which promotes histiocyte recruitment in ECD.
The overall prognosis for ECD depends on the severity of extra-skeletal lesions, such as pericardial tamponade and retroperitoneal involvement leading to renal failure from sepsis [3]. Overall 1-year and 5-year survival is approximately 96% and 68%, respectively [4]. Cardiovascular involvement confers an overall poor prognosis [4–6].
The diagnosis was possible due to the clinical pursuit of the aetiology of the pericardial effusion, and reinforces the importance of modern imaging in the diagnosis of a rare entity, raising the question of whether a body CT scan should be mandatory in the study of a pericardial effusion.