ABSTRACT
Acquired haemophilia is a bleeding disorder caused by antibodies against coagulation factors. Some cases are associated with autoimmune diseases. However, no cases of acquired haemophilia with eosinophilic fasciitis have been previously reported. Herein we describe the case of a patient with eosinophilic fasciitis associated with acquired haemophilia.
LEARNING POINTS
- Eosinophilic fasciitis is one of the underlying diseases of acquired haemophilia.
- The underlying disease of acquired haemophilia should be investigated thoroughly and systematically to optimize therapeutic strategies.
- This is the first report of acquired haemophilia in association with eosinophilic fasciitis.
KEYWORDS
Acquired haemophilia, eosinophilic fasciitis
INTRODUCTION
Acquired haemophilia is a rare, but often life-threatening bleeding disorder caused by autoantibodies, mostly against factor VIII (FVIII inhibitor) [1]. The incidence of the disease is estimated at approximately one to four cases per million population per year [1]. Although many cases are idiopathic (43.6–51.9%), some are associated with malignancy, pregnancy and autoimmune diseases (9.4–17.0%) [2]. Associated autoimmune diseases include rheumatoid arthritis, systemic lupus erythematosus, Sjögren’s syndrome, dermatomyositis, myasthenia gravis, Graves’ disease, giant cell arteritis, anti-neutrophil cytoplasmic antibody-associated vasculitis and Goodpasture’s syndrome, polyarteritis nodosa, and autoimmune haemolytic anaemia [3–10]. However, no cases of acquired haemophilia with eosinophilic fasciitis have been previously reported. Eosinophilic fasciitis, also called Shulman syndrome, is a rare connective tissue disease characterized by symmetrical erythema, oedema, and induration of the skin and soft tissues of the limbs and trunk [11, 12]. Extracutaneous involvement includes muscle pain and weakness, joint contractures (56%) and arthritis (40%) [11]. Laboratory evaluation often reveals eosinophilia and hypergammaglobulinemia. Serum levels of creatine kinase are typically normal, even in patients with myalgia. The aetiology of eosinophilic fasciitis is unknown and most cases are considered idiopathic. However, the following have been suggested as possible triggers or associated factors: strenuous exercise, haemodialysis, infections, some medications (statins, phenytoin, ramipril, heparin and immune checkpoint inhibitor), autoimmune diseases (thyroid disease, primary biliary cirrhosis, systemic lupus erythematosus and Sjögren’s syndrome) and haematological disorders (aplastic anaemia, acquired amegakaryocytic thrombocytopenia, myeloproliferative disorders, myelodysplastic syndromes, lymphoma, leukaemia and multiple myeloma) [11, 12]. Herein we describe the case of a patient with eosinophilic fasciitis associated with acquired haemophilia. To our knowledge, this is the first report of acquired haemophilia in association with eosinophilic fasciitis. Physicians should consider fasciitis as an underlying disease of acquired haemophilia.
CASE DESCRIPTION
A 79-year-old man who had a history of hyperlipidaemia and diabetes was referred because of a prolonged activated partial thromboplastin time (APTT) of 89.1 sec, decreasing haemoglobin (from 12.7 g/dl to 9.7 g/dl in 2 weeks) and oedema of the extremities. Physical examination showed purpura and oedema of the upper and lower extremities (Fig. 1).
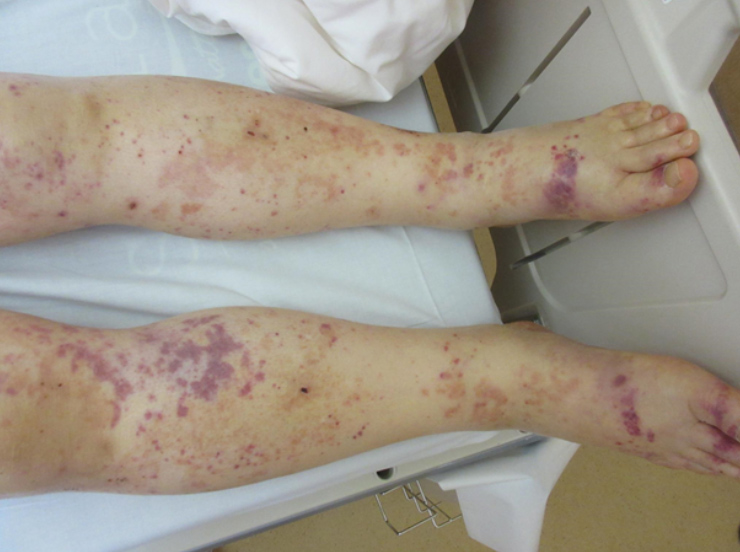
Figure 1 (click to enlarge)
Figure 1. A photograph of the patient’s skin showing purpura and oedema of the lower extremities
Skin sclerosis was not observed. No arthritis, muscle weakness, headache, nailfold capillary changes, digital ulcers or Raynaud’s phenomenon was present. Blood analysis showed anaemia (haemoglobin 9.7 g/dl), normal platelet count (30.3×104/µl), normal prothrombin time (prothrombin time–International Normalized Ratio 1.05), inflammatory syndrome (white blood cell count 9300/µl, C-reactive protein (CRP) 4.6 mg/dl, erythrocyte sedimentation rate (ESR) 84 mm/h) and allergic conditions (eosinophils 1647/µl, immunoglobulin E 4031 IU/ml; normal <170 IU/ml). APTT was prolonged (89.1 sec; normal <32 sec). FVIII level was decreased (1%; normal >60%). FIX and FXI levels were normal. APTT mixing studies showed the presence of circulating anticoagulant. Positive results were obtained for FVIII inhibitor antibody (5 Bethesda units/ml; normal <1.0 Bethesda units/ml). Tests for antiphospholipid antibody and lupus anticoagulant yielded negative results. Von Willebrand factor was normal. Rheumatoid factor (RF), anti-Sm antibody, anti-RNP antibody, anti-SS-A antibody and anti-neutrophil cytoplasmic antibody were all negative. Abdominal and thoracic computed tomography (CT) studies showed no evidence of malignancy or infection. Magnetic resonance imaging (MRI) of the extremities showed fascial thickening of the flexor muscles of the forearm and the brachioradialis, pronator teres, tibialis anterior and gastrocnemius muscles. Fascia and muscle biopsies were not performed due to the risk of uncontrolled bleeding.
Although there are no universally accepted diagnostic criteria, the Japanese Dermatological Association suggests the following [12]:
- Major criterion: symmetrical plate-like sclerotic lesions are present on the four limbs without Raynaud’s phenomenon and systemic sclerosis can be excluded.
- Minor criterion 1: the histology of a skin biopsy that incorporates the fascia shows fibrosis of the subcutaneous connective tissue, with thickening of the fascia and cellular infiltration of eosinophils and monocytes.
- Minor criterion 2: thickening of the fascia is seen using imaging tests such as MRI.
A definitive diagnosis is made when a patient has the major criterion and one of the minor criteria, or the major criterion and two of the minor criteria.
Our patient showed oedema of the upper and lower extremities and skin sclerosis was not observed (Major criterion). Also, MRI of his extremities showed thickening of the fascia (Minor criterion 1). Consequently, the patient was diagnosed with acquired haemophilia associated with fasciitis.
As no acute life-threatening bleeding was seen on admission, haemostatic therapy was not administered. Oral prednisolone was started at 40 mg/day (0.6 mg/kg). The clinical course is shown in Fig. 2. APTT immediately normalized and FVIII inhibitor antibody became negative in 2 weeks. Eosinophilia also immediately resolved. However, oedema of the upper and lower extremities remained unchanged. The prednisolone dose was increased to 70 mg/day (1 mg/kg). Oedema of the extremities diminished and follow-up MRI of the extremities showed improvement of fascial thickening for several muscles. On day 40, prednisolone was decreased to 40 mg/day without any subsequent relapses.
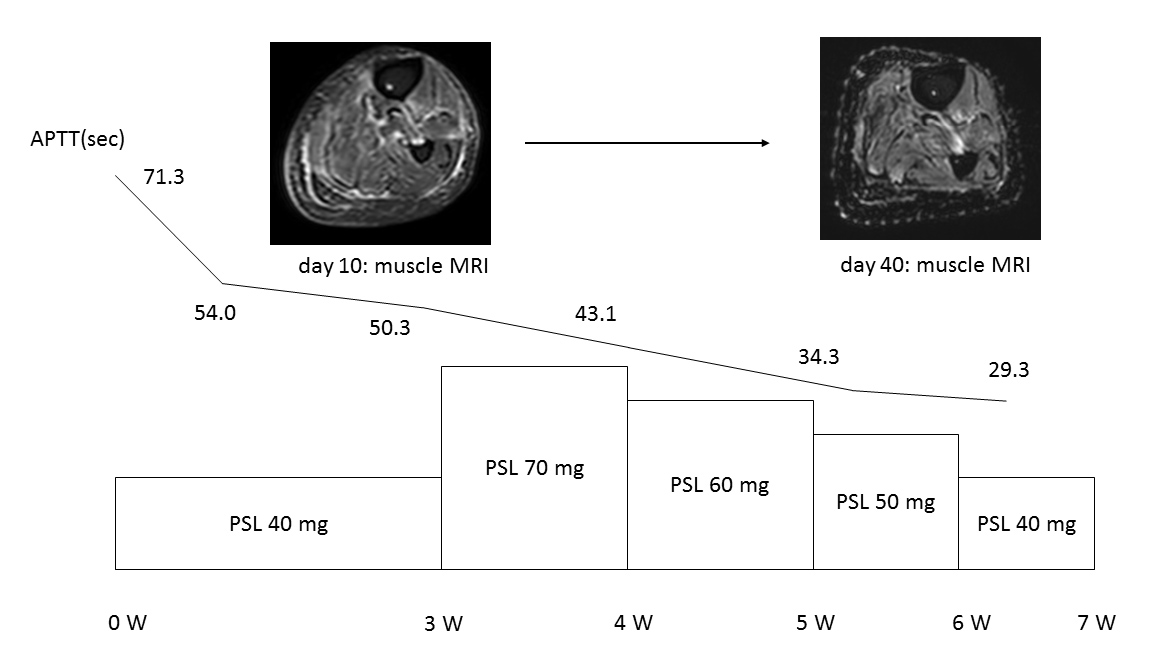
Figure 1 (click to enlarge)
Figure 2. The patient’s clinical course. Acquired haemophilia and eosinophilic fasciitis were sucessfully treated with corticosteroids. Muscle MRI studies showed fasciitis.
APTT, activated partial thromboplastin time; PSL, prednisolone; W, week
DISCUSSION
Acquired haemophilia is a rare, but often life-threatening bleeding disorder caused by autoantibodies, mostly against factor VIII (FVIII inhibitor). Some cases are associated with autoimmune diseases. We have provided the first description of acquired haemophilia in association with eosinophilic fasciitis. Physicians should consider fasciitis as an underlying disease of acquired haemophilia. In our case, the patient showed oedema of the upper and lower extremities and skin sclerosis was not observed. Also, although eosinophilic fasciitis could not be proved by muscle biopsy due to the risk of uncontrollable bleeding, an MRI study of the lower extremities showed fascial thickening of some muscles and eosinophilia was observed, consistent with the diagnostic criteria of the Japanese Dermatological Association. These findings strongly indicate the underlying disease of acquired haemophilia was eosinophilic fasciitis as some acquired haemophilias are associated with autoimmune diseases, although the possibility of the two diseases coincidentally occurring simultaneously could not be rule out.
Initial management of acquired haemophilia is to stop the acute bleeding. Recombinant or plasma-derived factor VIII, activated prothrombin complex concentrate, or recombinant factor VIIa is recommended [2]. In our case, as there was no acute bleeding on admission, haemostatic control was not necessary. On the other hand, as the factor VIII level was reduced (1%; normal >60%) and positive results were obtained for factor VIII inhibitor antibody (5 Bethesda units/ml; normal <1.0 Bethesda units/ml), it was important to eliminate factor VIII inhibitor. For this purpose, corticosteroids, immunosuppressants or a combination of these agents have all shown efficacy against acquired haemophilia[1]. However, optimal management methods have yet to be established due to the rarity of the disease, particularly in association with autoimmune diseases. Also, the mainstay of management for eosinophilic fasciitis is systemic corticosteroids. Although no randomized trials have evaluated therapies for eosinophilic fasciitis and the best approach to treatment remains unclear, initial management with systemic corticosteroids, usually starting at doses equivalent to prednisone 1 mg/kg per day, is recommended [13]. However, optimal management methods for eosinophilic fasciitis in association with other autoimmune diseases such as acquired haemophilia have yet to be established. In our case, prednisolone at 40 mg/day (0.6 mg/kg) was sufficient to normalized APTT and abolish FVIII inhibitor antibody. However, oedema of the upper and lower extremities remained unchanged. Prednisolone at 70 mg/day (1 mg/kg) reduced oedema of the extremities and improved the fascial thickening of several muscles. The dosage of prednisolone should be optimized. More case reports and large surveillance studies are required in order to clarify suitable therapeutic strategies.