ABSTRACT
New-onset systemic lupus erythematosus (SLE) is uncommon in elderly patients. We report the case of a 71-year-old woman who was diagnosed with SLE based on clinical manifestations of fever, alopecia, bicytopenia, hepatomegaly, lymphadenopathy, glomerulonephritis, positive antinuclear antibody (ANA) and anti-double stranded DNA (anti-dsDNA) antibody. Renal biopsy was consistent with lupus nephritis and excision biopsy of a right inguinal lymph node was initially reported as having features of reactive hyperplasia. However, a more careful review of the lymph node biopsy subsequently confirmed a concurrent angioimmunoblastic T-cell lymphoma. This case illustrates the importance of investigating secondary causes and possible alternative diagnoses in patients who present with atypical features of connective tissue disease, and the challenges in diagnosing a rare form of lymphoma.
LEARNING POINTS
- A thorough work-up for secondary causes and careful evaluation to exclude possible alternative diagnoses is important in cases of elderly-onset lupus.
- The disease presentations of lupus and haematological malignancies such as lymphoma may mimic each other and differentiation between the two can be clinically challenging; lupus can be associated with cytopenias, hepatomegaly and lymphadenopathy, but the degree of severity and the context of the clinical presentation need to be considered carefully before attributing these features to it.
- As some lymphomas are rare and difficult to diagnose, if there is a high clinical suspicion despite negative histological studies, discussion with the pathologist is important and a review of histology should be sought.
KEYWORDS
Elderly-onset systemic lupus erythematosus, lupus nephritis, angioimmunoblastic T-cell lymphoma
CASE DESCRIPTION
A 71-year-old woman with a background of hypertension, hyperlipidaemia and osteoarthritis presented to hospital with fever for 1 day. She reported having alopecia for the last 4 years with no other symptoms. Blood pressure was elevated at 157/73 mmHg on admission. On examination, she was noted to have mild hepatomegaly and palpable inguinal lymph nodes. No other abnormalities were found on examination of her cardiovascular, respiratory, abdominal and neurological systems. She also did not exhibit any other features of connective tissue disease such as malar rash, oral ulcers or synovitis.
Initial laboratory investigations revealed bicytopenia with a non-haemolytic anaemia and leucopenia. Serum creatinine was elevated (86 µmol/l; reference range 44–88) and urine studies showed microscopic haematuria with dysmorphic predominance, and proteinuria of 2.26 g/day. Serum albumin was 34 g/l and other liver function tests were unremarkable.
Blood cultures were negative, along with tests for tuberculosis (TB; TB DNA by ProbeTec, acid-fast smear and culture) on lymph node and bone marrow specimens. Dengue testing (by IgM antibody and dengue virus NS1 antigen), Epstein–Barr virus (EBV) PCR and parvovirus B19 PCR testing were negative. Cytomegalovirus (CMV) IgM antibody was equivocal, and PCR testing subsequently came back negative. Hepatitis B and C, and human immunodeficiency virus (HIV) screens were also negative.
Antinuclear antibody titre was positive (ANA, >1:640, homogeneous pattern) along with raised anti-double stranded DNA antibody titres (anti-dsDNA, 324.63 IU). Serum complement levels were low (C3 0.43 g/l; reference range 0.45–83, C4 0.03 g/l; reference range 0.11–0.41 g/l). In view of the positive ANA and anti-dsDNA serologies, low complement levels, history of alopecia, bicytopenia and glomerulonephritis, a diagnosis of systemic lupus erythematosus (SLE) was made. A renal biopsy was carried out which helped to confirm the diagnosis of SLE. It showed a focal mesangial proliferative glomerulonephritis with global and segmental sclerosis, consistent with class II to very mild class III disease (Fig. 1).
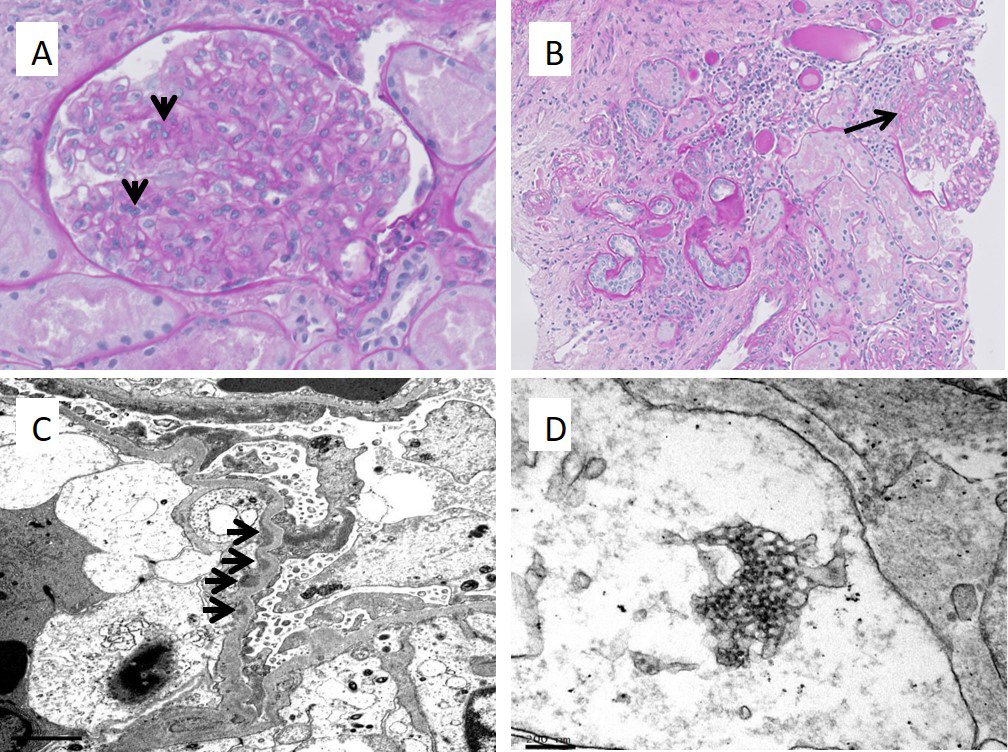
Figure 1 (click to enlarge)
Figure 1. Renal biopsy images showing features consistent with lupus nephritis. (A) PAS stain demonstrating segmental mesangial hypercellularity (4-to 5 cells, arrows), with few swollen endothelial cells and circulating mononuclear cells. (B) PAS stain showing a glomerulus with a small segment of sclerosis (arrow). Patchy tubular atrophy with several interstitial lymphocytes and plasma cells are noted. (C) Electron micrograph shows extensive podocyte foot process effacement. There are small subendothelial electron dense deposits (arrows). (D) A tubuloretical structure is seen on electron microscopy
As the onset of SLE is uncommon after the age of 50 years, additional testing to exclude other possible alternative diagnoses was carried out. A computed tomography scan of the thorax, abdomen and pelvis showed multiple intra-abdominal and intra-thoracic lymphadenopathies (the largest measuring 1.6 cm in the short axis in the right inguinal region) with hepatomegaly of 17.3 cm. Excision biopsy of the right inguinal lymph node was subsequently performed and histological analysis was initially reported as having features of reactive hyperplasia. However, in view of a high clinical suspicion of malignancy, a more careful review of the histology was sought, which confirmed a concurrent angioimmunoblastic T-cell lymphoma (AITL), mostly Attygalle pattern 1 with focal pattern 2 (Figs. 2 and 3. Bone marrow aspiration and trephine biopsy did not show lymphomatous involvement.
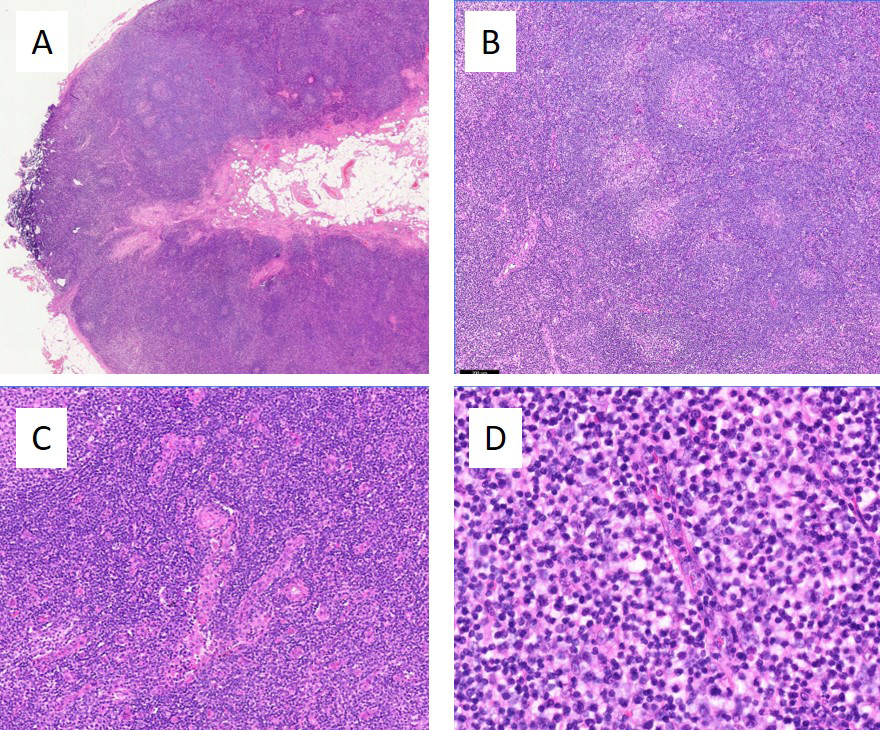
Figure 2 (click to enlarge)
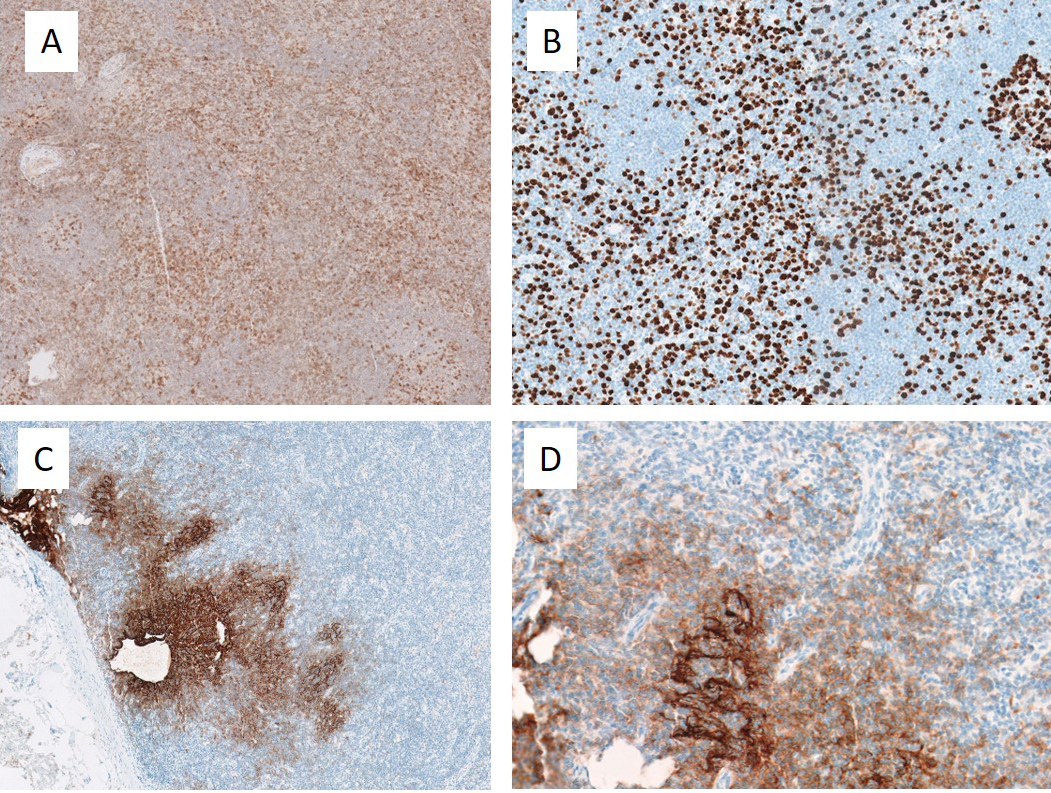
Figure 3 (click to enlarge)
Figure 2. Lymph node biopsy, H&E stains. (A) On low magnification, the lymph node showed apparent architectural preservation, with circumscribed germinal centres, patent subcapsular sinuses and no extranodal extension. (B) However, on closer inspection, the architecture appared somewhat distorted and altered. B-follicles, while rounded, showed hyperplastic and atretic germinal centres and poorly defined mantle zones. (C) The paracortical regions were expanded by small to intermediate sized lymphocytes with associated hypervascularity. (D) At higher magnification, the paracortical lymphocytes showed irregular nuclear contours and clear cytoplasm
Figure 3. Lymph note biopsy. Immunohistochemical stains showed that the paracortical lymphoproliferation comprised an atypical T-cell population expressing pan-T cells antigens CD2, CD3 and CD5, with minor downregulation of CD7 and an increased proliferation index up to 50%. They also showed excessive extrafollicular expression of CD4, iCOS, and to lesser extent PD-1 and CD10, consistent with a T-follicular helper phenotype. CD21 demonstrated that some follicular dendritic meshworks were irregularly expanded, with tentacular enwrapment around paracortical high endothelial venules. (A) iCOS, low magnification. Increased expression is seen outside of germinal centres. (B) Ki67, low magnification. The interfollicular proliferation fraction is aberrantly increased. A residual germinal centre is present at the top right. (C) CD21, low magnification. Follicular dendritic meshworks show irregular expansion. (D) CD21, high magnification. Follicular dendritic meshworks show tentacular enwrapment around the endothelial venules
After diagnosis of the patient’s elderly-onset lupus, prednisolone 60 mg once daily (equivalent to 1 mg/kg/day) and hydroxychloroquine were initiated while the results of further investigations were awaited. When AITL was confirmed, the patient was offered treatment with the CHOP (cyclophosphamide, doxorubicin, vincristine and prednisolone) regimen for the treatment of her lymphoma but she declined all forms of chemotherapy. Therefore, a decision was made to start her on cyclosporin 50 mg twice daily instead. Cyclosporin is known to have some activity against AITL and would also help to treat her SLE (with predominant haematological involvement and lupus nephritis). However, in view of worsening renal function and transaminitis after she was started on cyclosporin, it was held off and she was continued on prednisolone. With treatment, there was complete resolution of her fever, haematuria and proteinuria and stabilization of her creatinine at 138 µmol/l (reference range 44–88 µmol/l). A subsequent follow-up CT scan demonstrated an interval reduction in size of her lymph nodes. Prednisolone was subsequently tapered to 5 mg once daily and she remains clinically well at the time of writing.
DISCUSSION
The main differential diagnoses considered in our patient were SLE, malignancy and systemic infection. She fulfilled the diagnosis of SLE according to the 2012 SLICC (Systemic Lupus Erythematosus International Collaborating Clinics Group) criteria based on her clinical presentation, and laboratory and renal histological findings. An infectious screen as detailed did not reveal any underlying infection. However, the diagnosis of her concurrent angioimmunoblastic T-cell lymphoma was a challenging one.
Rheumatological disorders have long been associated with cancers [1], and some atypical features of clinical presentation have raised the suspicion of an underlying malignancy, such as the onset of lupus in an elderly patient, prominent hepatomegaly and extensive lymphadenopathy. Given these clinical findings, it was more important to rule out haematological malignancy.
AITL is uncommon, representing 1–2% of all non-Hodgkin lymphoma. It is difficult to diagnose on histology and is often under-recognized with delayed diagnoses common [2]. The cell of origin for AITL is the follicular T helper cell (TFH), a type of CD4+ T cell that regulates antibody responses. As such, dysregulated TFH function in AITL can often lead to autoimmune manifestations. The early stages of AITL with Attygalle pattern 1 or 2 may mimic a reactive lymph node due to apparent architectural preservation at lower magnification and polymorphous reactive infiltrate. Often, a high index of suspicion and experience is needed to make the diagnosis.
In the case of our patient with elderly-onset lupus with widespread lymphadenopathy, lymphoma was a key differential diagnosis. In addition, hepatomegaly is also not a common presentation in lupus (5% at onset, and up to 25% at any time) [3], and usually occurs in the context of hepatic steatosis or hepatitis [4, 5]. As there were no features to suggest these conditions and suspicion for an underlying malignancy was high, the case and lymph node histology were discussed again with a haematopathologist. This helped determine the concomitant diagnosis of AITL in this patient.