ABSTRACT
Background: Sickle cell disease is a genetic condition frequently found in Africa and the Arabian Peninsula. Uncommon complications include subgaleal haematoma (soft head syndrome) and periorbital oedema.
Case presentation: A 17-year-old male patient presented with body aches and progressive right parieto-temporal and frontal head swelling. Physical examination revealed puffiness of the right eye that progressed rapidly to reddish periorbital oedema sparing the extraocular muscle and pupil response to light. CT and MRI of the brain suggested multiple subgaleal haematomas (soft head syndrome) and right periorbital oedema.
Conclusion: Subgaleal haematoma (soft head syndrome) and periorbital oedema are uncommon complications of sickle cell disease. Management is conservative rather than surgical.
LEARNING POINTS
- Subgaleal haematoma concurrently with periorbital oedema is a rare presentation of sickle cell disease.
- There are no guidelines on treatment, but the conditions in our patient resolved with conservative management.
KEYWORDS
Sickle cell, subgaleal hematoma, periorbital edema, soft head syndrome, orbital compression syndrome
INTRODUCTION
Sickle cell disease (SCD) is found in Africa and the Arabian Peninsula. Disease burden is higher in Africa due to environmental and genetic factors, but the Arabian Peninsula has various genotypes, haplotypes and phenotypes. Disease presentation is milder in the Arabian Peninsula but heterogeneous, with some patients having a severe clinical course and complications [1]. In 2001, a review of five SCD cases in Oman described orbital wall infarction which presents acutely during vaso-occlusive crisis [2]. A rare complication of SCD is subgaleal haematoma, with a few cases reported sporadically, some of them in Saudi Arabia [3, 4].
CASE PRESENTATION
A 17-year-old Saudi male patient with SCD and speech difficulty presented to our centre. He had no history of ICU admission, cerebrovascular accident, acute chest syndrome or thrombosis. However, since birth he has been admitted to hospital once or twice a year for simple blood transfusions. There is no history of alloimmunization or exchange transfusion. He presented to our centre via the emergency room with a 4-day history of headache and bilateral temporal swelling which progressed downwards to the frontal area, then the periorbital area and then the right side of his face. There was no history of trauma, fever or neurological symptoms. This was his first time to develop such a presentation.
On examination, the patient was stable and afebrile with a temperature of 36.7°C. His blood pressure was 128/74 mmHg, pulse rate was 80 bpm, respiratory rate was 20 per minute, and oxygen saturation was 98–100% on room air. His face was swollen, particularly on the right side. There was right periorbital swelling with mild redness over the upper eyelid. The swelling was not warm or tender but pitting in nature over the forehead. Eye movement was intact and normal. The sclera was slightly jaundiced but there was no redness (Fig. 1). The rest of the systemic examination was unremarkable. There were no neurological deficits and all cranial nerves, motor function, sensation and gait were intact.
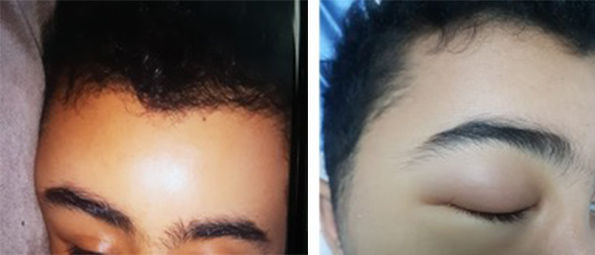
Figure 1 (click to enlarge)
Figure 1. (Left image) Right lateral frontal swelling and eye oedema at presentation to the emergency room. (Right image) Complete right eye closure with peri-orbital oedema a few hours after presentation.
Initial laboratory studies showed haemoglobin 9.23 g/dl, haematocrit 26%, white blood cell count 5.39×103/µl, platelet count 126×103/µl, and lactate dehydrogenase 337 U/l. The liver function test showed indirect hyperbilirubinemia (total bilirubin 36 µmol/l, conjugated bilirubin 9.09 µmol/l). Baseline haemoglobin electrophoresis revealed HbS 78% and HbF 11%. C-reactive protein was 225 mg/l and a renal function test was normal.
The patient was provisionally diagnosed with periorbital oedema/haematoma with cellulitis and was commenced on intravenous amoxicillin-clavulanate and IV fluids.
A computed tomography scan (CT) of the head showed diffuse soft tissue oedema of the scalp which was more pronounced on the right side with extension to the right preseptal region. A few subgaleal collections were noted in the right frontoparietal, right anterior frontal, bilateral high frontal and left posterior parietal regions with thin peripheral enhancement, not invading the underlying skull bone. Content attenuation reached 26 HU and the largest collection measured 8.4 (length) ×1.3 (thickness) cm (Fig. 2). The differential diagnosis consisted of inflammatory fluid collections (subperiosteal abscesses) and acute soft head syndrome (bony infarcts with subperiosteal haematomas).
On the third day of admission, the patient’s haemoglobin dropped to 7.87 g/dl and he was transfused with one unit of packed RBCs with haemoglobin rising to 9.5 g/dl after transfusion.
On the fourth day of admission, MRI of the brain showed multiple fluid collections in the right frontal and high parietal regions bilaterally, likely subperiosteal in location with haemorrhagic content, as well as underlying skull bone abnormal intensities, raising the possibility of soft head syndrome rather than an infectious process (Fig. 3). The diffuse soft tissue oedema that was noted on a previous CT scan of the head was no longer seen.
By the fifth day of admission, the patient’s facial oedema had improved dramatically compared with admission day, with no right periorbital swelling or conjunctival redness and only slight swelling on the right cheek. At a follow-up appointment in the haematology clinic 1 week later, the patient had no complaints and the cheek swelling had resolved (Fig. 4). A repeat CT scan of the brain 4 weeks later showed complete interval resolution of the previously noted diffuse soft tissue oedema and multiple subgaleal collections.
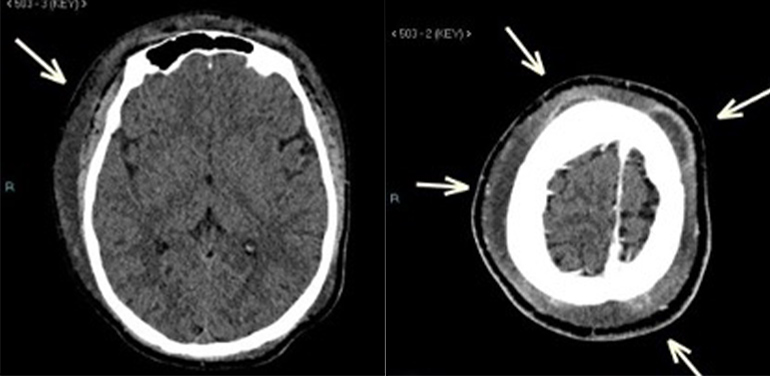
Figure 2 (click to enlarge)
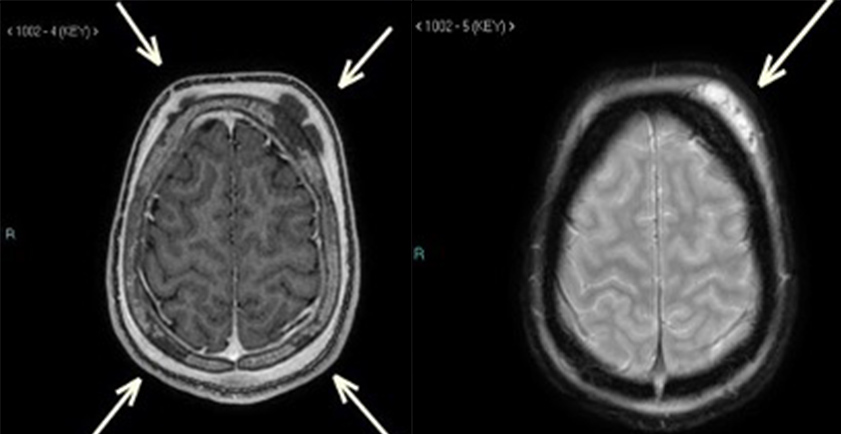
Figure 3 (click to enlarge)
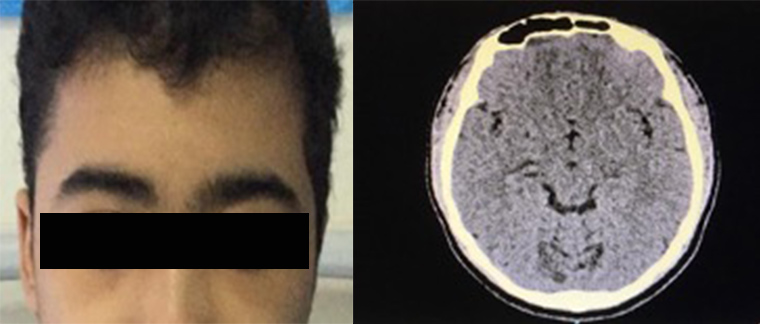
Figure 4 (click to enlarge)
Figure 2. CT scan of the brain at presentation. (Left image) Plain study showed diffuse soft tissue oedema of the scalp which was more noted on the right side. (Right image) Post-contrast study showed subgaleal collections in the frontoparietal areas bilaterally.
Figure 3. MRI of the brain. (Left image) T1-weighted post-contrast image showed multiple fluid collections in the frontoparietal regions bilaterally, likely sub-periosteal in location with haemorrhagic content, as well as underlying skull bone intensities, raising the possibility of soft head syndrome. (Right image) T2-weighted image showed a left frontal sub-periosteal collection
Figure 4. (Left image) Patient`s face after discharge with full recovery, and no frontal head swelling or periorbital oedema. (Right image) CT scan of the brain after discharge showed complete resolution of the previous subgaleal collection
DISCUSSION
SCD is a genetic haemoglobin disorder inherited in an autosomal recessive manner. It was first identified in the eastern region of Saudi Arabia in the 1960s. It is a common disorder in Saudi Arabia with significant variations in prevalence across the country. The eastern region has the highest prevalence rate, followed by southern and south-western areas [5]. As in other countries, SCD in Saudi Arabia is associated with high mortality and morbidity.
Patients with SCD can present with haemolytic, sequestration or aplastic crisis, or more commonly with vaso-occlusive or pain crisis. Vasoconstriction, decreased blood volume and bone or organ infarction underlie all crises. Bone infarction is more common in long bones than in skull bone; some cases of skull bone infarction have been reported but the incidence has not been determined [6]. As the disease becomes more chronic, there are more complications with intra- and extramedullary haematopoiesis.
Subgaleal haematoma is caused either by head trauma or by a non-traumatic cause, so-called spontaneous subgaleal haematoma or soft head syndrome. Non-traumatic causes include iatrogenic injury, coagulopathy, arterio-venous fistulas or ruptured aneurysms of a superficial artery [7]. Subgaleal haematoma is a rare complication of SCD. Although it is not properly understood, subgaleal haematoma might be caused by repeated bone infarction with decreased blood supply to the periosteum resulting in elevation of the periosteum and cortical bone destruction [8]. However, Dahdaleh et al. have suggested that in SCD the chronic disease process with increased medullary haematopoiesis affects skull bone anatomy causing spontaneous subgaleal haematoma or acute soft head syndrome [9].
Sporadic cases of subgaleal haematoma in SCD have been reported in the literature. Foula et al. described one case of spontaneous subgaleal haematoma in an SCD patient. They also presented a review of 32 cases with spontaneous subgaleal haematoma and/or epidural haematoma; only 12 cases were pure subgaleal haematoma with no epidural haematoma [3]. Three of the cases were treated by craniotomy, one received unknown therapy and the remaining eight were treated conservatively [3].
Ophthalmological manifestations and orbital involvement in SCD as a result of vaso-occlusive crisis have been described but are uncommon. The patient can present with glaucoma, retinopathy, retinal vessel occlusion, anterior segment ischaemia, angioid streaks or with a more serious condition such as orbital compression syndrome (OCS) [10]. Periorbital oedema is rare and may be part of OCS due to auto-infarction of the orbital bones. In addition to eyelid oedema, OCS is characterized by proptosis and periorbital pain, which was not seen in our patient[11]. MRI is the radiological modality of choice to evaluate orbital changes.
Ganesh et al. described five different presentations of periorbital oedema in SCD. No patient had subgaleal haematoma, two had proptosis and in all MRI showed soft tissue changes in the orbital area. All patients were treated with antibiotics and steroids as well as simple or exchange transfusion [2]. Our patient had isolated periorbital oedema with no ocular pain or proptosis and with normal MRI findings (Fig. 5). This presentation suggests premature OCS, simple isolated periorbital oedema as a separate orbital manifestation of SCD, or extension of subcutaneous oedema from reactive changes associated with soft head syndrome.
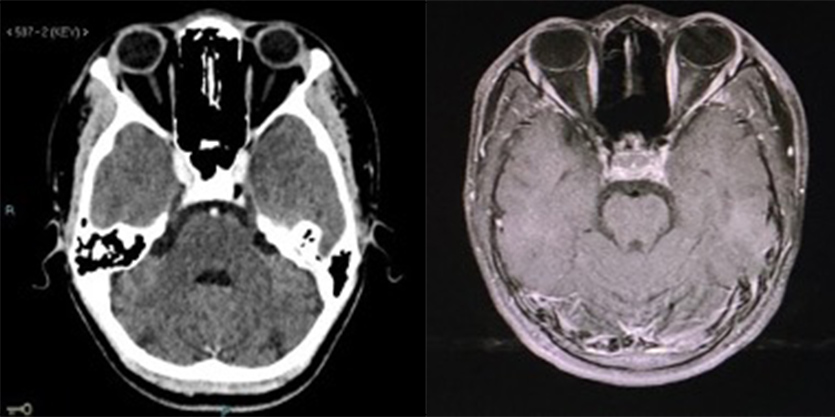
Figure 5 (click to enlarge)
Figure 5. (Left image) CT scan of the brain showed a normal orbital cavity. (Right image) MRI of the brain showed no periorbital collection or proptosis
In conclusion, subgaleal haematoma (soft head syndrome) and periorbital oedema are uncommon complications of SCD with uncertain pathophysiology. The combination of two rare conditions in the same patient presented a challenge as we are unaware of any other similar presentations and there are no guidelines for treating subgaleal haematoma and/or periorbital oedema in SCD. However, conservative management with good hydration and treatment to reduce haemoglobin S resulted in a favourable outcome. Nevertheless, more research is needed in this area.