ABSTRACT
Objectives: To describe the diagnosis and treatment of a severely ill patient presenting with thrombotic microangiopathy (TMA) of unknown cause.
Case presentation: An adult female presented to intensive care with abdominal pain and haemorrhagic shock, requiring reanimation.
Results: Features of TMA were present, but initial plasma exchange was ineffective. Treatment with the anti-C5 antibody, eculizumab, improved laboratory parameters and organ function, albeit slowly. Eculizumab remains effective and well tolerated after 30 months of treatment.
Conclusion: This case demonstrates the complexities and importance of early identification of atypical haemolytic uraemic syndrome in patients presenting with TMA.
LEARNING POINTS
- It is important to establish the underlying cause of thrombocytopenia, haemolytic anaemia and organ failure quickly due to the rapid and severe progression of the disease.
- Multiple organs may be affected in aHUS and on presentation, no identification of STEC and normal ADAMTS13 activity are strong indications for a suspicion of aHUS (with or without a known abnormality of the complement system).
- In this case, eculizumab treatment quickly resolved clinical status and, progressively, the laboratory parameters. Despite the delayed diagnosis, renal function improved with eculizumab although the patient still has stage 3B chronic kidney disease, highlighting the importance of rapid diagnosis and appropriate management.
KEYWORDS
Chronic kidney disease, eculizumab, haemolytic uremic syndrome
INTRODUCTION
Thrombotic microangiopathy (TMA) is a severe and quickly progressing condition with different causes. Since choice of management varies, rapid differentiation of disorders in which TMA can be a complication, and those defined by TMA, such as haemolytic uraemic syndrome (HUS) and thrombotic thrombocytopenic purpura (TTP), should be attempted in order to initiate the most appropriate treatment as quickly as possible. HUS is divided into Shiga toxin-producing Escherichia coli HUS and the rarer atypical HUS (aHUS), which is caused by uncontrolled complement activation. Distinguishing between these forms is essential[1].
CASE PRESENTATION
Presentation and diagnosis
An adult female (born 1951) was admitted to intensive care in January 2012 having experienced bloody diarrhoea, nausea and vomiting 6 days prior to hospitalization. The patient had abdominal pain, was in haemorrhagic shock due to gastrointestinal haemorrhage from diffuse gastritis and required reanimation. She exhibited features of TMA: thrombocytopenia (89×109/l, normal range 140–360×109/l), acute kidney injury with elevated serum creatinine (18.6 mg/dl, normal range 0.5–0.9 mg/dl), elevated lactate dehydrogenase (LDH; 2432 U/l, normal range <247 U/l) and low haemoglobin (7.9 g/dl decreasing to 4 g/dl, normal range 12–16 g/dl).
ADAMTS13 (a disintegrin and metalloproteinase with thrombospondin motifs 13) testing showed enzyme activity was 49% and antigen levels (0.52 µg/ml) were within normal range (0.50–1.60 µg/ml), but the patient tested positive for antibodies (19 IE/ml). Retesting 2 weeks later revealed a negative antibody result. There was no sign of malignancy or increased erythropoiesis upon bone marrow smear and the patient tested negative for Shiga toxin-producing Escherichia coli (STEC) and disseminated intravascular coagulation (DIC).
Treatment
The patient immediately received intensive plasma exchange (PE) with 4 l of plasma (15–17 bags daily) and was on artificial ventilation for the first 10 days following a hypertensive crisis (220/120 mmHg) with lung oedema. She developed critical illness polyneuropathy and pneumonia, sepsis due to the insertion of a Sheldon catheter, and had several hypertensive crises. Due to acute kidney failure, dialysis was performed daily for 16 days, then every other day for a further 7 weeks. Rituximab (650 mg) weekly for 4 weeks was started during the third week in the ICU, and during the fifth week, plasma volume was increased to 6 l (24 bags) daily. Although levels of LDH had improved to 236 U/l, creatinine (2.9 mg/dl), haemoglobin (6.6 g/dl) and platelets (74×109/ml) remained outside normal ranges (Fig. 1A).
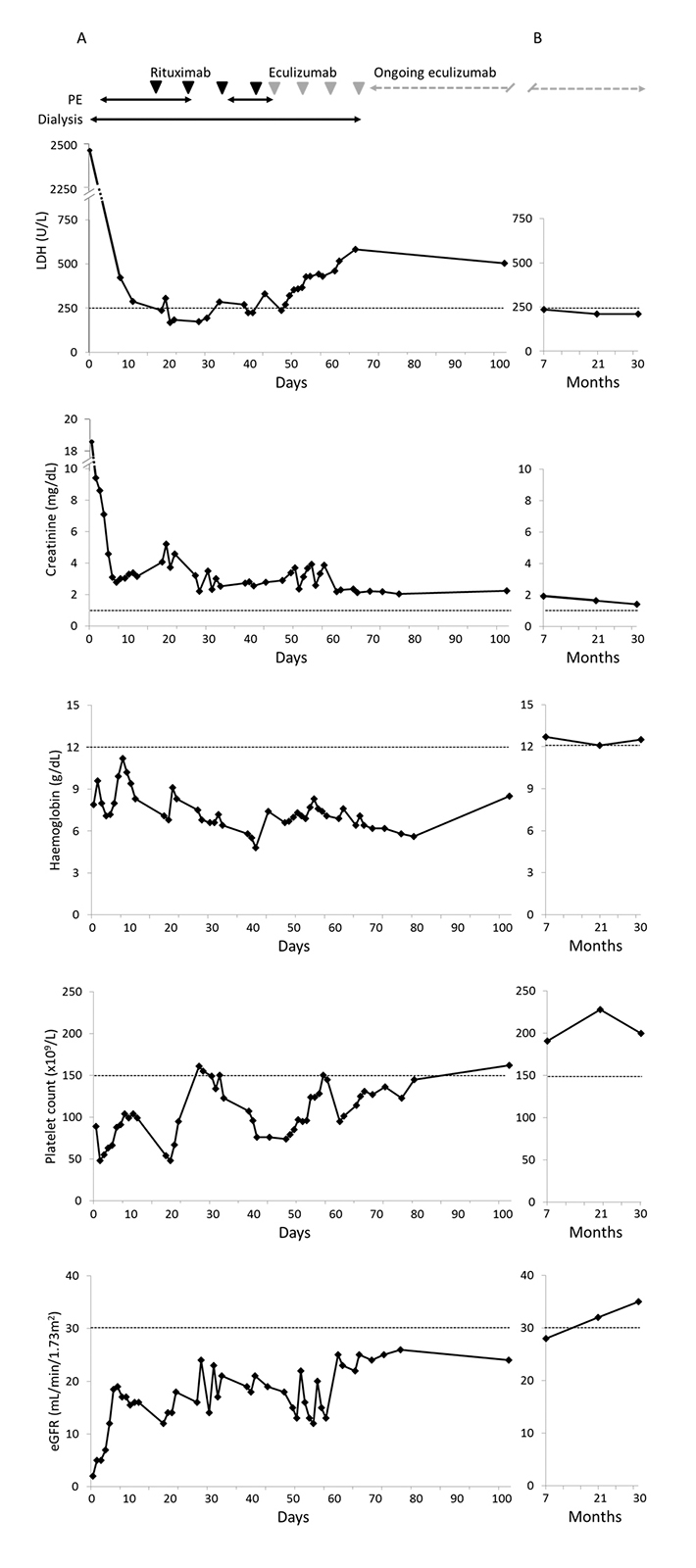
Figure 1 (click to enlarge)
Figure 1: Management of an adult female with aHUS, as demonstrated by levels of LDH, creatinine, haemoglobin and platelet count and estimated glomerular filtration rate (eGFR) (A) during an initial period of hospitalization and (B) at follow-up of 7, 21 and 30 months. The horizontal axis represents time since admission to hospital. Dashed lines represent the upper normal limit (LDH and creatinine) or lower normal limit (haemoglobin and platelets) or severely reduced renal function: stage 3 CKD (eGFR).
In the absence of a satisfactory response to treatment, reconsideration of the symptoms led to the diagnosis of aHUS, an extremely rare condition. Therefore, PE and rituximab were discontinued and the approved treatment for aHUS, eculizumab, was initiated (900 mg weekly for 4 weeks followed by maintenance therapy of 1200 mg every 2 weeks). Before starting eculizumab therapy, the patient was vaccinated against Neisseria meningitidis.
Outcome and follow-up
On eculizumab treatment, platelet count and estimated glomerular filtration rate (eGFR) increased (Fig. 1A). The patient was moved to a normal ward after the second dose of eculizumab. Clinical and laboratory parameters were initially slow to improve, but dialysis was discontinued after 3 weeks of eculizumab and she was discharged after 2 months of eculizumab treatment (4 months after initial admission); platelet level was normal (162×109/l) and eGFR was 24 ml/min/1.73 m2 (Fig. 1). Complement activity (C3 and C4) was tested and found to be low. Seven months after presentation, a kidney biopsy revealed no signs of TMA; however, mild tubular injury was noted. The patient experienced a hypertensive crisis after the renal biopsy. The patient was followed up 30 months after presentation (July 2014) whilst still receiving eculizumab. Platelet count (200×109/l), LDH (210 U/l) and haemoglobin (12.5 g/dl) were within normal ranges (Fig. 1B). Renal function had improved, with creatinine levels just above normal (0.5–0.9 mg/dl) at 1.4 mg/dl and eGFR of 35 ml/min/1.73 m2 (CKD stage 3B).
DISCUSSION
This case highlights the difficulties in diagnosing and recognizing aHUS, a rare, complex and severe condition characterized by uncontrolled complement activation causing TMA and subsequent organ damage. On presentation, TMA, organ impairment, absence of STEC and normal ADAMTS13 activity are all strong indications for a diagnosis of aHUS, but as it is a rare disorder, it was not initially considered. Severe gastrointestinal involvement may complicate diagnosis due to its association with STEC-HUS, and it is thus essential to report when present in patients with aHUS. It is important to distinguish these forms of HUS, as eculizumab has shown significant efficacy in aHUS in prospective trials, while only anecdotal evidence shows efficacy in STEC-HUS[2].
This case also emphasizes the importance of early and specific treatment of aHUS to avoid continuing and irreversible complement-mediated TMA damage to the kidneys (and potentially at extrarenal sites such as the central nervous and cardiopulmonary systems). The initiation of PE and supportive care was of limited efficacy in this case, as the underlying cause was not targeted. Once eculizumab was initiated, the patient was moved from the ICU to a normal ward, PE and dialysis were discontinued and haemolysis progressively resolved. This was accompanied by improved renal status. Laboratory parameters initially improved slowly, but the patient’s clinical situation rapidly improved and effective long-term control was demonstrated at 30 months of follow-up. This sustained resolution of TMA with eculizumab is in line with previous studies[3,4].
The renal recovery with eculizumab treatment in this patient is worthy of note. On discharge, the patient had stage 4 chronic kidney disease; at follow-up, levels of creatinine remained near normal, eGFR had increased to 35 ml/min/1.73 m2, and a renal biopsy found no signs of TMA. A similarly impressive degree of renal recovery has also been reported recently in a case of eculizumab therapy after >3 months of dialysis[5], but is not seen in all cases if treatment with eculizumab is initiated too late[6]. Our case indicates that eculizumab therapy is of clinical benefit despite delayed diagnosis of aHUS and established kidney failure, an outcome also reported by Povey et al.[5].
The long-term effects of eculizumab treatment are still being established. Our patient experienced no major side effects during 30 months of therapy, and long-term treatment of aHUS with eculizumab was well tolerated in a prospective study of patients with aHUS[4].
SUMMARY
This case highlights the importance of early diagnosis of this rare and severe disease of TMA that can affect multiple organ systems and adds to the body of evidence on the tolerability and effective use of eculizumab in the treatment of aHUS. As well as normalization of haematological parameters, there is increasing evidence that what is often considered established renal disease can be improved with eculizumab therapy.