ABSTRACT
Pheochromocytoma, papillary thyroid carcinoma and hyperparathyroidism have rarely been reported together. Whether this association is coincidental or results from an unknown genetic predisposition is difficult to ascertain.
We present the case of a patient who was diagnosed with pheochromocytoma, bilateral papillary thyroid carcinoma and parathyroid hyperplasia with primary hyperparathyroidism. A genetic mutation was hypothesized as the connection between these lesions. Previously described mutations were explored.
LEARNING POINTS
- Parathyroid hyperplasia, primary hyperparathyroidism and papillary thyroid carcinoma individually are common conditions, but association with each other, although possibly incidental, should trigger genetic testing.
- Further research is needed to reliably explain the relationship between primary hyperparathyroidism and non-medullary thyroid cancer.
KEYWORDS
Pheochromocytoma, hyperparathyroidism, papillary thyroid carcinoma
INTRODUCTION
Pheochromocytomas occur most commonly as sporadic tumours, but approximately 35–40% of cases are inherited as part of distinct syndromes such as von Hippel Lindau syndrome (VHL), multiple endocrine neoplasia type 2 (MEN 2), neurofibromatosis type 1 or paraganglioma syndrome. In MEN 2, there is an association between medullary thyroid carcinoma, pheochromocytoma and primary hyperparathyroidism. However, papillary thyroid carcinoma is rarely associated with pheochromocytoma, and the relationship between these two diseases remains unclear [1].
CASE DESCRIPTION
We report the case of a 59-year-old woman, with a known history of pheochromocytoma who presented to our department for the evaluation of a multinodular goitre.
The patient had previously presented at 52 years of age with a hypertensive crisis associated with headache, flushes and palpitations. Urinary catecholamine values were elevated and the abdominal ultrasound revealed a large heterogenic mass measuring 38×38 mm in the right adrenal gland. The CT scan showed a right adrenal mass measuring 45×40 mm. 131I-metaiodobenzylguanidine (131I-MIBG) scintigraphy showed increased uptake in the right adrenal gland without distant metastases. Right adrenalectomy was performed with a favourable postoperative outcome. Pathological examination confirmed the diagnosis of pheochromocytoma.
Screening for MEN revealed a nodular goitre with bilateral nodules. Thyroid function tests, calcitonin and the PTH level were normal. The pentagastrin-stimulated calcitonin test was performed and was negative. The patient continued follow-up for her nodular goitre, which remained stable. Screening for pheochromocytoma relapse was negative every year.
When the patient was 59 years of age, the thyroid ultrasound showed significant enlargement of the nodules in the right thyroid lobe. Thyroid scintigraphy revealed a cold nodule in the left lobe. Fine needle aspiration showed a benign follicular lesion. Thyroid function tests and calcitonin were again normal, but the serum calcium level was at the upper limit of the normal range (2.5 mmol/l, NV 2.15–2.5, albumin 41 g/l), with an elevated parathyroid hormone level (138 pg/ml, NV 14–72), increased calciuria (394.8 mg/24 h, NV 100–3000, calcium/creatinine ratio 0.305 mg/mg, NV 0.2 mg/mg), despite a normal 25OH vitamin D level (35.3 ng/ml) and normal renal function (GFR >90 ml/min), confirming the diagnosis of primary hyperparathyroidism. Abdominal ultrasonography did not revealed nephrolithiasis or nephrocalcinosis; osteoporosis was confirmed by osteodensitometry (L1–L4 BMD 0.741 g/cm², T –2.6, Z –1.2; right hip BMD 0.900 g/cm², T –0.2, Z=0.2, forearm BMD 0.612 g/cm², T –1.4, Z –0). Parathyroid scintigraphy with sestamibi was negative (Fig.1).

Figure 1 (click to enlarge)
Figure 1. SPECT images are difficult to interpret because of a large goitre. Several bilateral intrathyroidal uptake areas can be seen
A computed tomography scan showed a multinodular goitre with net compression of the trachea. It was decided that a total thyroidectomy with intraoperative parathyroid exploration should be performed (Fig. 2). The thyroid was removed as were the two right parathyroid glands, together with the superior left gland, because of their hyperplastic appearance.
Pathology examination showed bilateral papillary carcinoma with follicular presentation and vascular invasion (TNM classification pT3N×M×V1) (Fig. 3). Thyroid tumour cells were negative for calcitonin and CEA. Parathyroid glands were hyperplastic. I-131 ablation therapy (100 mCi) was administered after surgery.
The patient was screened for germline mutations in a panel of candidate genes: RET (exons 8, 10, 11, 13, 14, 15 and 16), VHL, SDHB, SDHC, SDHD and SDHA, which turned out to be negative. A genetic study using next generation sequencing searching for 50 genes linked to malignancy in thyroid tumours was performed. A mutation was found in HRAS (mutation pG13R in exon 2). The family history of this patient was negative.
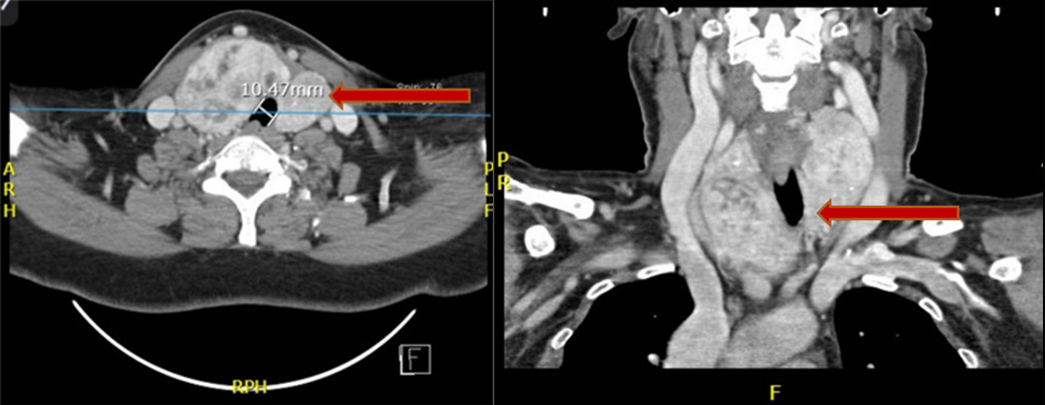
Figure 2 (click to enlarge)
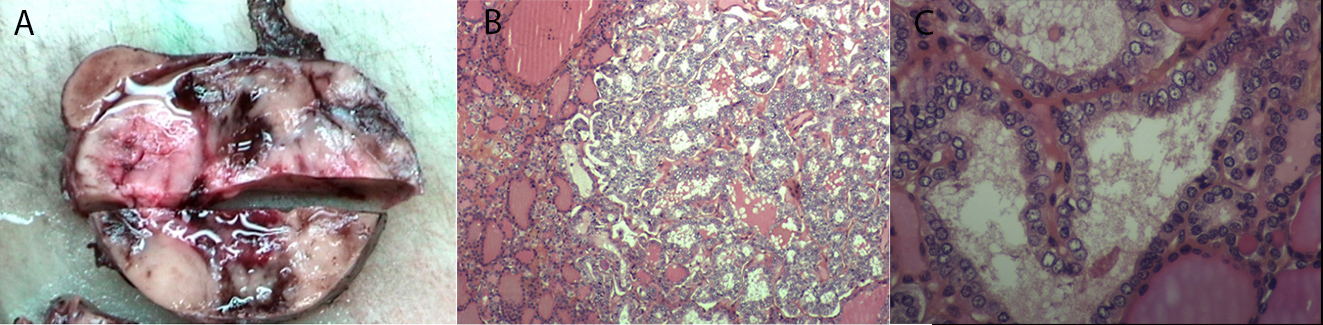
Figure 3 (click to enlarge)
Figure 2. CT scanning shows a multinodular goitre with deviation and compression of the trachea
Figure 3. Anatomopathological examination. (A) Macroscopic: papillary carcinoma with follicular presentation, measuring 6 cm, with vascular invasion.
(B and C) Microscopic: proliferation with a microfollicular architecture; the follicles are formed of cells with large, pale and atypical nuclei
DISCUSSION
We have described an unusual association of pheochromocytoma, bilateral papillary thyroid carcinoma and parathyroid gland hyperplasia with primary hyperparathyroidism in a patient with an unrevealing family history. This combination of endocrine disease could not be explained by any syndrome or conceivable interrelationships among the tumours.
The almost invariable component of MEN, medullary thyroid carcinoma, was absent in our patient. Furthermore, the genetic study was negative for the most frequently involved genes in these three pathologies. Only a HRAS mutation was found in the thyroid tumour, which is generally associated with the follicular variant of papillary thyroid carcinoma.
The incidence of papillary thyroid carcinoma in patients with pheochromocytoma seems to be greater than in the general population. Multiple theories may explain this. Fluctuating thyrotropin secretion caused by circulating catecholamine may be a contributing factor[1]. Pheochromocytomas can produce and release peptides, such as insulin-like growth factor II and hypothalamic-like or pituitary-like hormones that may stimulate the development and/or growth of papillary thyroid carcinoma [2].
Although primary hyperparathyroidism and simultaneous medullary thyroid disease is well described in the literature as a part of MEN 2 syndrome, the coexistence of hyperparathyroidism and papillary thyroid cancer has only been documented in sporadic case reports and in some small series.
The mechanisms underlying the relationship between primary hyperparathyroidism and papillary thyroid cancer have not yet been established. However, an increased risk of non-parathyroid cancers in patients with primary hyperparathyroidism has been reported in several studies. Miccoli et al. [3] reported thyroid malignancy as the most prevalent cancer among patients with primary hyperparathyroidism. While some authors suggested this was coincidental, others investigated several predisposing factors such as exposure to neck radiation and increased mitotic activity induced by hypercalcemia [4]. In addition, the tumour-promoting effect of parathyroid hormone and subsequent genetic predisposition to new malignancies was also noted [5]. Further research is still needed to reliably explain the relationship between primary hyperparathyroidism and non-medullary thyroid cancer.
On the other hand, we know that encapsulated and infiltrative follicular variants of papillary thyroid carcinomas have different genotypic profiles. The encapsulated follicular variant has a BRAF and RAS mutation pattern very similar to follicular adenoma/carcinoma (with RAS+++), whereas the infiltrative follicular variant has a BRAF and RAS genotype lying between follicular adenoma/carcinoma and classic papillary carcinoma (BRAF+++) [6]. Our patient had a papillary cancer with vascular invasion, with a RAS mutation while testing for the BRAF mutation was negative. However, the functional consequence of this RAS mutation is unknown.
CONCLUSIONS
This is a report of an unusual association of pheochromocytoma, bilateral papillary thyroid carcinoma and primary hyperparathyroidism with parathyroid hyperplasia. The relationship between these conditions remains unclear in this case; it may be a coincidental association or might result from genetic variants still awaiting identification.