ABSTRACT
Rendu-Osler-Weber syndrome is a rare inherited syndrome with autosomal dominant transmission characterized by systemic arteriovenous malformations (AVMs) with multi-organ involvement. Its incidence is 1–2/100,000 and it is predominant in females (the male/female ratio varies from 1:2 to 1:4.5).Clinical manifestations and complications are related to recurrent bleeding and, in some cases, the development of end-organ failure. Management is mostly supportive care and it is essential to promote control of the disease as much as possible and screen eventual complications.
We describe the case of a 67-year-old male patient with Rendu-Osler-Weber syndrome admitted to the emergency department with decompensated heart failure due to acute anaemia because of severe epistaxis. During hospitalization, the patient progressed to acute-on-chronic liver failure with hepatic encephalopathy and an abdominal computed tomography scan showed multiple hepatic AVMs considered to be the cause of the chronic liver disease.
LEARNING POINTS
- Rendu-Osler-Weber syndrome is a rare autosomal dominant syndrome characterized by systemic arteriovenous malformations (AVMs) with multi-organ involvement, in which the most common manifestation is recurrent epistaxis.
- In more severe cases the prognosis is determined by organ dysfunction caused by AVMs, including hepatic involvement, which happens in 74–79% of cases, leading to poor outcomes.
- In more severe cases the prognosis is determined by organ dysfunction caused by AVMs, including hepatic involvement, which happens in 74–79% of cases, leading to poor outcomes.
KEYWORDS
Rendu-Osler-Weber syndrome, arteriovenous malformations, epistaxis, congestive heart failure, chronic hepatic disease
CASE DESCRIPTION
We present a case of a 67-year-old male patient with a medical history of Rendu-Osler-Weber syndrome with severe recurrent epistaxis requiring frequent blood transfusions and chronic therapy with tranexamic acid. Other relevant diagnoses included diastolic heart failure of unclear aetiology with multiple hospitalizations due to cardiac decompensation, atrial fibrillation with no possibility of anticoagulation therapy due to haemorrhagic risk, chronic liver disease with no definitive aetiology and iron deficiency anaemia. The patient had 2 brothers and 2 sons, all of whom were diagnosed with Rendu-Osler-Weber syndrome, none with major complications. It is unknown whether the patient or family members underwent a genetic study.
The patient was admitted to the emergency department with a new episode of decompensated heart failure in the context of anaemia (haemoglobin of 5.5 g/l) caused by severe epistaxis and needed transfusion support. An echocardiogram showed left ventricular dilatation, preserved global systolic function (ejection fraction of 54%), right cavity overload, flattening of the interventricular septum resulting in a D-shaped left ventricle, biauricular dilatation and a dilated right ventricle with good function. Despite an echocardiogram suggestive of pulmonary thromboembolism, it was decided not to proceed with the investigation due to the impossibility of performing anticoagulation therapy.
During hospitalization, the patient experienced periods of greater sleepiness with other periods of psychomotor agitation and mental confusion. Blood tests revealed hyperbilirubinaemia with total bilirubin 21.4 mg/dl, direct bilirubin 13.7 mg/dl, gamma-glutamyltransferase 74 IU/l and alkaline phosphatase 114.74 IU/l, consistent with acute-on-chronic liver failure with hepatic encephalopathy. An abdominal computed tomography scan showed multiple arteriovenous malformations (AVMs) between the hepatic artery and the suprahepatic veins and heterogeneity due to numerous micronodular and hypervascular lesions related to vascular malformations and/or millimetric haemangiomas (Fig. 1).
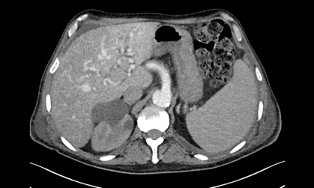
Figure 1 (click to enlarge)
Figure 1. A abdominal CT scan documented multiple arteriovenous malformations between the hepatic artery and the suprahepatic veins and heterogeneity due to numerous micronodular and hypervascular lesions related to vascular malformations and/or millimetric haemangiomas
These liver AVMs are a manifestation of Rendu-Osler-Weber syndrome and were the probable cause of the chronic liver disease. The systemic arteriovenous shunts and the chronic anaemia could also explain secondary high-output heart failure. During the subsequent hospital stay, the clinical condition deteriorated, with new episodes of epistaxis and hepatic encephalopathy with no response to lactulose enemas. The patient died 3 weeks after hospital admission.
DISCUSSION
Rendu-Osler-Weber syndrome, also named hereditary haemorrhagic telangiectasia (HHT), is a systemic fibrovascular dysplasia characterized by AVMs [1,2]. Telangiectasias (small AVMs) mostly affect the lips, oropharyngeal mucosa, face, chest and fingers, though they can also be found in the pulmonary, hepatic and cerebral circulations, but the most common clinical manifestation is spontaneous and recurrent epistaxis, which is typically the initial sign of disease, frequently in childhood.
This disorder is the result of mutations in 2 genes, one coding for endoglin (ENG), located on chromosome 9q33-q34, and another coding for activin receptor-like kinase 1 (ALK1), located on chromosome 12q13. The proteins modulate transforming growth factor β (TGF-β), which stimulates vascular endothelial growth factor (VEGF) production and plays a key role in angiogenesis. ENG-related mutations are observed in HHT type 1, the phenotype responsible for 40% of pulmonary AVMs. Mutations affecting ALK1 are observed in HHT type 2, leading to lower incidence of these AVMs (14%), associated with milder forms and late onset of the disease [2–4].
The diagnosis is based on 3 or more of the subsequent clinical features: spontaneous and recurrent epistaxis, multiple telangiectasias at typical locations (lips, oropharyngeal mucosa, face, fingers), visceral involvement of the AVMs (such as pulmonary, hepatic, cerebral) and family history (a first-degree relative with HHT) [2]. Two of these criteria define the case as suspected Osler-Weber-Rendu syndrome. Identification of a heterozygous pathogenic variant in ACVRL1, ENG, GDF2 or SMAD4 genes establishes the diagnosis if clinical features are inconclusive [5].
Visceral AVMs are normally asymptomatic; however, complications from bleeding or shunting may be abrupt and catastrophic, especially when they occur in the lungs, liver or brain [5]. Severe complications such as haemorrhagic stroke in cerebral AVMs, high-output heart failure and liver cirrhosis in hepatic AVMs and paradoxical embolic stroke or brain abscess in pulmonary AVMs [2] may occur. The mortality rate due to disease complications is less than 10% of cases [2]. Organ involvement is determined by the genotype. Pulmonary and cerebral AVMs are more common in HHT1 patients, while hepatic AVMs and hepatic AVM-associated pulmonary hypertension are usually found in those with HHT2 [2,3].
The prevalence of hepatic involvement ranges from 74–79% [1]. Hepatic manifestations of HHT include arteriovenous shunts, arterioportal shunts and portovenous shunts that can lead to high-output heart failure. Chronic liver ischaemia also occurs and can lead to fibrosis and liver cirrhosis. Screening for hepatic AVMs is recommended even in asymptomatic individuals with HHT1 using hepatic Doppler ultrasound, CT or MRI as second-line techniques. An echocardiogram should be performed to evaluate the haemodynamic repercussions in the heart and lungs [1].
Treatment of this disorder is only supportive, the objectives being to relieve symptoms, manage complications and prevent progression of the disease [2].
Although most cases are asymptomatic or with minor complications, with the exception of recurrent epistaxis, it is essential to carry out an extensive investigation to identify the location of the AVMs and the systemic repercussions of this disorder. Therefore, it is important to have early recognition of hepatic, cerebral and pulmonary AVMs and a close follow-up to prevent the development of severe complications, actions which are associated with a more favourable prognosis [1].
In our clinical case in particular, where all 4 diagnostic criteria were present, prognosis was adversely determined by the late diagnosis of hepatic AVMs, which led to end-organ failure such as heart and liver failure, ultimately resulting in the patient’s death.