ABSTRACT
Primary aortic sarcoma is a rare and aggressive malignancy with only approximately 190 cases reported in the literature. While angiosarcoma and intimal sarcomas represent an estimated 67.7% of malignant aortic tumours, spindle cell sarcomas are even more exclusive, consisting of only 0.9% of malignant aortic tumours. Differentiated from other malignant aortic tumours, spindle cell sarcomas are of mesenchymal origin and usually express vimentin and osteopontin. Clinical presentations are variable and nonspecific, ranging from back pain, abdominal pain or elevated blood pressure, misleading to differentials like pulmonary emboli or aortic aneurysms such as in our case here. In this article, we discuss the finding of an extremely rare aortic sarcoma masquerading as a pulmonary embolism. The patient underwent surgical resection; however, the course was complicated by the development of brain metastases and intracranial haemorrhage. The literature is expanding regarding the evolution of adjuvant chemotherapy and radiation therapy in the treatment of these patients. The exact pathogenesis of spindle cell sarcomas is unknown but thought to be related to the MDM2-p53 pathway. The development of spindle cell sarcomas may be related to Li-Fraumeni syndrome, which should be on the differential for these patients. This case highlights the importance of identifying aortic sarcomas in patients who present with signs and symptoms of peripheral embolization as the diagnosis can be easily misconstrued for thrombus or aortic aneurysm, leading to a delay in proper and timely management. We herein emphasize that aortic sarcomas should be included in the clinician’s working differential due to the poor prognosis and outcomes that these aggressive tumours carry.
LEARNING POINTS
- Malignant aortic tumours are rare and can present with a multitude of symptoms ranging from constitutional symptoms to abdominal discomfort to unexplained hypertension. Spindle cell sarcomas represent 1 of the least common malignant aortic tumours reported in the literature.
- Malignant aortic tumours have a poor prognosis, and of the various types of malignant aortic tumours, aortic sarcomas have a particularly poor prognosis with a 5-year survival rate of 8%.
- The exact pathophysiology of these malignancies is unknown but is thought to be related to the MDM2-p53 pathway and may be related to Li-Fraumeni syndrome.
KEYWORDS
Primary aortic sarcoma, malignant aortic tumour, spindle cell, pulmonary embolism, Li-Fraumeni syndrome
INTRODUCTION
Primary aortic sarcoma is a rare and aggressive entity, which includes leiomyosarcoma, fibrosarcoma, haemangioendothelioma, myxoid sarcoma and angiosarcoma [1]. There are only approximately 190 cases reported in the literature [2]. These can originate from different parts of the aorta including the descending thoracic aorta (34.9%), abdominal aorta (27.3%), thoracoabdominal aorta (26.5%) and the aortic arch (11.3%) [2]. Clinical manifestations of primary aortic tumours are variable and nonspecific, including mostly constitutional symptoms (32.1%), abdominal complaints (28.5%), aneurysm or pseudoaneurysm (26.7%), back pain (22.4%) or hypertension (18.2%) [3]. The clinicopathologic classification by Wright et al. divides aortic sarcoma into intimal and mural types [4], while the immunohistochemical pattern proposed by Thalheimer et al. classifies intimal angiosarcomas, intimal myofibroblastic sarcomas and mural sarcomas [5]. These aggressive tumours are associated with poor outcomes and have a mere estimated 5-year survival rate of 8% [6]. The poor prognosis of these patients is mostly attributed to tumour-related complications, such as infarction from tumour embolization or ostial occlusion, as well as metastatic disease to bone, liver, kidney and/or skin [6]. Due to the worldwide sparsity of cases of aortic tumours, knowledge pertaining to their diagnosis and management, including the evolving role of adjuvant chemo- and radiation therapy, remains limited. Herein, we present a rare case of a patient with high-grade spindle cell sarcoma involving the intima of the aorta.
CASE DESCRIPTION
A 47-year-old Caucasian female with a history of fibromyalgia and diverticular disease initially presented to the emergency department with 1 week of progressively worsening shortness of breath on exertion, left-sided chest and back pain and dark bowel movements of 2 weeks’ duration. She was admitted with an initial diagnosis of symptomatic anaemia secondary to presumed gastrointestinal bleeding. Upon examination, she was tachypnoeic to 26 breaths/minute, hypertensive to 166/94 mmHg and had generalized pallor. The physical examination was otherwise unremarkable. Laboratory tests on admission included a complete blood count, a comprehensive metabolic panel, troponin and faecal occult blood tests. The results were significant for a haemoglobin of 5.8 mg/dl and a positive faecal occult blood test. The patient received 2 units of packed red blood cells and was admitted to the hospital. During her hospital stay, oesophagogastroduodenoscopy was performed and showed gastritis and duodenitis with no active bleeding. An intravenous proton pump inhibitor was started without improvement in her symptoms. Subsequent testing to rule out pulmonary embolism and aortic dissection included computed tomography angiography of the chest that was negative for pulmonary embolism, but revealed a large, unexpected focal abnormality of the anterior ascending aorta of uncertain aetiology. No overt dissection of the aorta was visualized. Further evaluation of the aorta included magnetic resonance imaging of the chest, which demonstrated a confluent hyperintense area of soft tissue surrounding the margins of the ascending thoracic aorta, arising superior to the aortic root and extending to the proximal aortic arch, and measuring 5.1×5.6×5.8 cm with mass effect compressing the superior vena cava (Fig. 1).
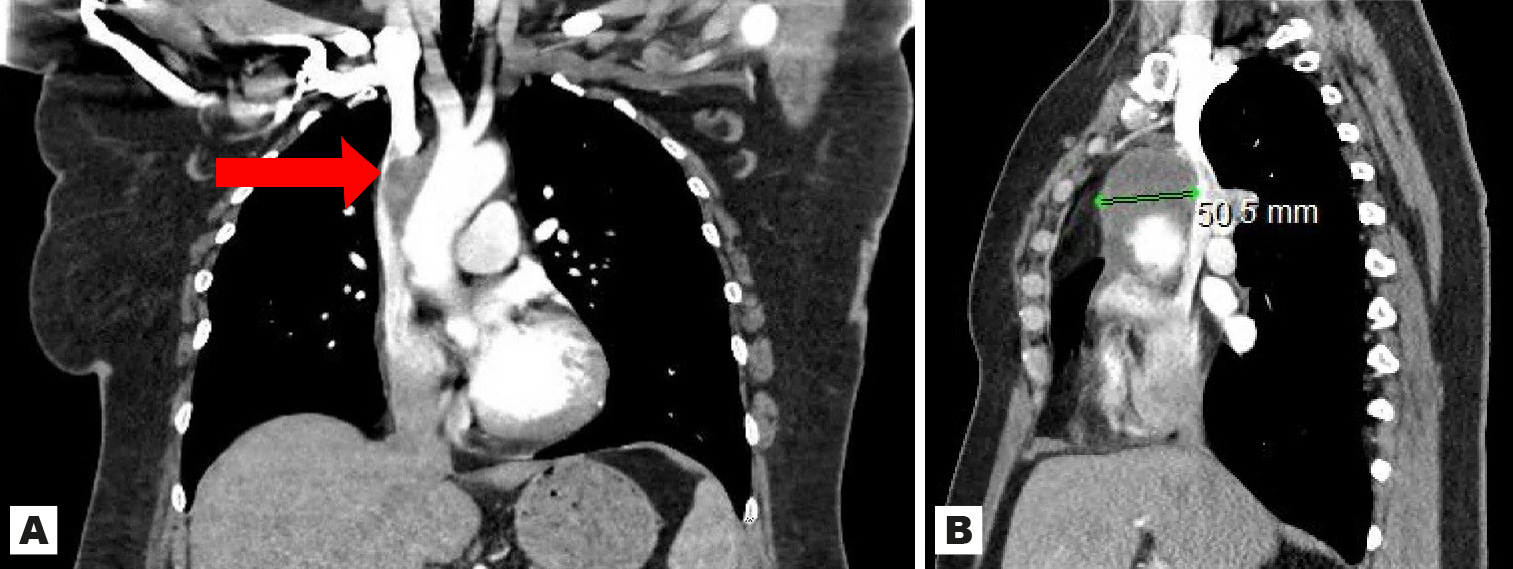
Figure 1 (click to enlarge)
Figure 1. Large soft tissue mass (arrow) intricately associated with the ascending thoracic aorta, arising superior to the level of the aortic root and proximal aortic arch with 90% mass effect on the superior vena cava identified on magnetic resonance imaging of the chest. Coronal view (A) and sagittal view (B)
Heart function in the setting of a newly identified proximal aortic mass was assessed via a transthoracic echocardiogram showing a mildly enlarged left ventricle, an ejection fraction of 55%, normal function of the aortic and mitral valves and no pericardial effusion. At this time, the patient remained symptomatic with documented evidence of a large ascending aortic mass with superior vena cava compression. Therefore, the decision was made to proceed with aortic repair and mass resection. Prior to operative management, an elective right and left heart catheterization demonstrated normal coronary anatomy and heart function. The patient then underwent successful ascending aortic replacement with a 24 mm graft and excision en masse of the superior anterior mediastinal mass contiguous with the aortic arch. An intra-operative transoesophageal echocardiogram demonstrated proper aortic graft placement and continued normal cardiac function. Initial frozen section pathology assessment of the periaortic mass demonstrated a spindle cell neoplasm arising from the aortic wall with sarcoma features. Further pathology assessment demonstrated atypical spindle cells in intersecting fascicles with increased mitotic activity and large areas of necrosis. These findings were consistent with a high-grade spindle and pleomorphic sarcoma (undifferentiated pleomorphic sarcoma) that was directly involving the intima and extending into adjacent soft tissue (Fig. 2).
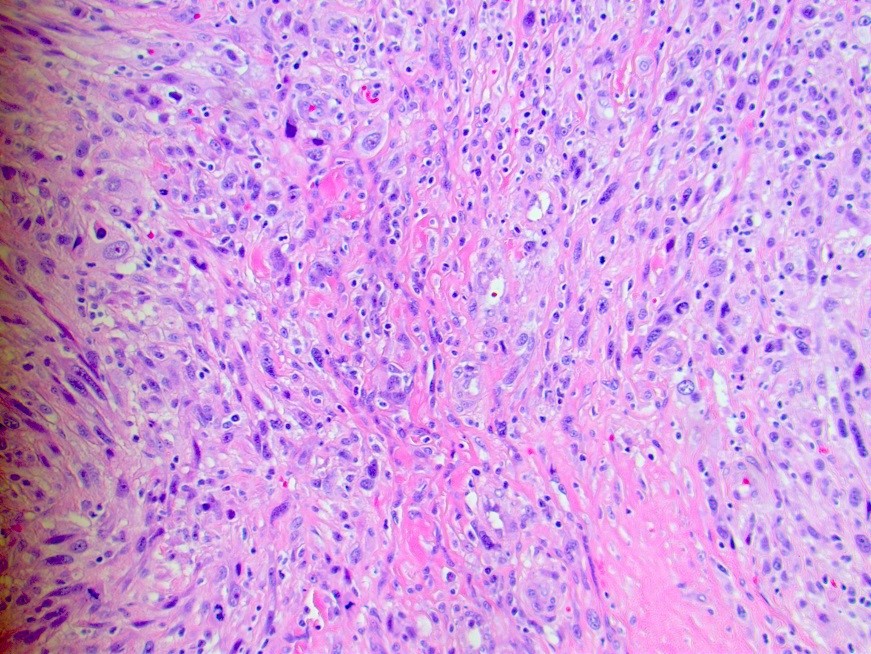
Figure 2 (click to enlarge)
Figure 2. Biopsy of the periaortic mass demonstrated atypical spindle cells in intersecting fascicles with increased mitotic activity and large areas of necrosis, consistent with high-grade spindle and pleomorphic sarcoma. Tumour cells were negative for CDK4 and MDM2 expression. Ki67 staining showed an increased proliferation rate of 40–50%
Immunohistochemical analysis showed that the tumour cells were negative for SMA, desmin, muscle-specific actin, CD34, CAM 5.2, CD3, CD20 and S100. Ki67 staining showed an increased proliferation rate at 40–50%. Additional immunohistochemical stains performed at Memorial Sloan Kettering Cancer Center showed that the tumour cells were negative for CDK4 and MDM2 expression, suggesting against an intimal sarcoma. Proximal and distal resection margins were negative for tumour. The patient tolerated the procedure well and was then discharged home on post-operative Day 5 with outpatient GI follow-up for a colonoscopy and oncology for a follow-up PET-CT scan, which showed hypermetabolic activity associated with the right thyroid gland as well as a left adrenal nodule. The patient was to receive systemic chemotherapy with doxorubicin (Doxil) for 4 cycles followed by pazopanib (Votrient) when she unfortunately presented to a different facility several months later for persistent, dull frontal headaches and was found to have multiple intracranial haemorrhagic masses in the right side of the brain with transtentorial herniation and midline shift (Fig. 3). She subsequently underwent right temporal craniotomy for resection of tumour. The pathology was consistent with high-grade spindle and undifferentiated pleomorphic sarcoma and compared to the previous pathology report, appeared to be the same tumour. One week postoperatively, the patient was sent to a rehabilitation institute for 5 days and was subsequently discharged home in a stable condition pending follow-up with radiation oncology and oncology.
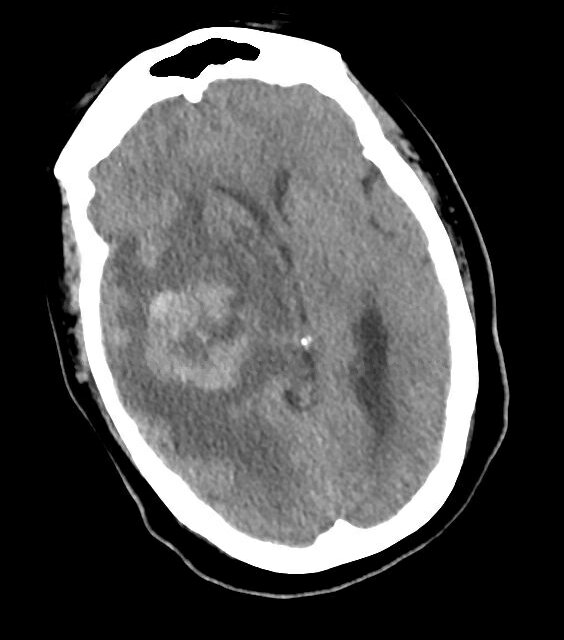
Figure 3 (click to enlarge)
Figure 3. Haemorrhagic metastasis with surrounding oedema and mass effect (causing right to left shift) on the right midbrain identified on head CT without contrast
DISCUSSION
Malignant aortic tumours are rare malignancies that hold a poor prognosis, with a median survival of 8 months after diagnosis [7].
Malignant aortic tumours can affect any segment of the aorta, with the descending thoracic aorta being affected in 34.9% of cases, the thoracoabdominal aorta affected in 26.5% of cases, the abdominal aorta affected in 27.3% of cases and the aortic arch involved in 11.3% of cases [2]. There are several different types of malignant aortic tumours, and the most common types include angiosarcoma and intimal sarcoma, representing an estimated 67.7% of malignant aortic tumours [7]. The least common aortic tumours are spindle cell sarcoma and myxofibrosarcoma, representing a mere estimated 0.9% and 0.4% of malignant aortic tumours, respectively [7].
Malignant aortic tumours have a slight predominance in the male sex and are usually present in middle-aged patients [8]. It is uncertain what constitutes a risk factor for developing malignant aortic tumours. In a review of 223 cases of malignant aortic tumours by Vacirca et al., 48.8% of patients had hypertension, 25.3% were smokers and 9.3% had type 2 diabetes mellitus; however, these risk factors are not clearly defined in the literature [7].
Along with being rare, malignant aortic tumours are also difficult to diagnose – in the study by Vacirca et al. 19.8% of patients were diagnosed post-mortem [7]. Malignant aortic tumours pose a challenge to diagnose since 1) they can present in a multitude of ways, and 2) they have no specific imaging characteristics [2]. On imaging, malignant aortic tumours are commonly confused with atherosclerotic disease [9]. One way in which malignant aortic tumours can be distinguished from atherosclerotic disease is by their extension into the vessel wall, but this can be difficult to appreciate [1]. Although it can be a difficult diagnosis to make, it is imperative to have a pre-operative diagnosis since late diagnosis can lead to incomplete resection of the tumour [9].
Classically, malignant aortic tumours present with signs of peripheral embolization, which may have a variety of symptoms, including intestinal ischaemia, claudication and/or neurological changes [9,10]. Of note, presentation with signs of peripheral embolization has been found to be a poor prognostic factor [7]. Many patients also present with back pain, as with our patient, leading to an initial presumptive diagnosis of an aortic aneurysm [7]. An estimated 29.1% of patients present with asthenia and 10.3% present with fever, which are suggestive of malignancy but are also nonspecific and limited in their diagnostic aid [7]. Patients with malignant aortic tumours do not typically present with signs of heart failure, although patients with cardiac tumours have been reported to present with acute heart failure, presumably due to the difference in location [10,11]. Furthermore, patients have been reported to present with cutaneous metastatic lesions, pain from metastasis and subclavian steal syndrome [7,9,12].
There are several modes of classification of malignant aortic tumours, such as by the location of origin [13]. Sarcomas that originate in the intima are called intimal sarcomas while those that originate in the media or adventitia are called mural sarcomas [13]. Differentiation between intimal and mural sarcomas can be made via immunohistochemical markers, such as CD31. MDM2 overexpression is found in 70% of intimal sarcomas and is thought to be a poor prognostic marker [9]. The gold standard of diagnosis remains immunohistochemical analysis [9,14]. Our patient was diagnosed with a type of intimal sarcoma called spindle cell sarcoma. Spindle cell sarcoma is one of the least reported tumours in the literature [15]. It originates in the connective tissue and can occur in any organ [15]. Spindle cells are not specific to spindle cell sarcoma or even intimal sarcomas, as they are also seen in other malignancies, such as synovial sarcomas [10]. Spindle cell sarcoma also has MDM2 overexpression, although this is not a specific feature of spindle cell sarcoma [10]. Spindle cell sarcomas are of mesenchymal origin and usually express vimentin and osteopontin [11]. The exact pathogenesis of spindle cell sarcoma is unknown but the supposed mechanism involves changes in the MDM2-p53 pathway [9]. Interestingly, a connection has been made between spindle cell sarcomas and Li-Fraumeni syndrome, an autosomal dominant syndrome that confers a predisposition to malignancy linked to a germline mutation of the TP53 gene, a tumour suppressor gene [16]. This particular gene mutation causes loss of function of p53, leading to downstream effects permissive of various types of cancer throughout life. Li-Fraumeni syndrome component tumours most commonly include osteosarcoma (12.6%), brain tumours (12%) and soft tissue sarcomas (11.6%). Of the sarcomas less frequently reported in the literature, spindle cell sarcomas and undifferentiated pleomorphic sarcomas are included [17]. Although our patient did not undergo genetic testing or have a known positive family history of early-onset cancer, Li-Fraumeni syndrome should be a diagnostic consideration in patients with aortic spindle cell sarcomas.
Given that malignant aortic tumours, particularly spindle cell sarcoma, are so rare, there are presently no universal guidelines for treatment[11]. However, surgery remains the mainstay of treatment, while the benefits of chemotherapy and radiation are unclear [11]. The most effective treatment is resection with graft interposition, but this is often not possible due to late patient presentation and anatomical considerations[9]. Thus, due to the rarity of these tumours, there is not as yet a proven role for adjuvant chemotherapy or radiation therapy in the treatment of these patients.
CONCLUSION
In conclusion, we present an extremely rare clinicopathological entity. Due to their aggressive nature and rarity, aortic sarcomas remain a challenge in terms of diagnosis and management. We thus suggest that aortic sarcoma should be suspected in patients with symptoms of distal embolic events, as this aggressive malignancy with grim prognosis requires prompt recognition and treatment.
This case demonstrates the non-specific nature porphyria could present with. Abdominal pain could easily have been dismissed as related to hepatitis and there are several causes of blistering disorders. This shows that despite being rare, a diagnosis of porphyria should be included in the differential diagnosis when appropriate since this has potentially life-saving implications for the patient and their relatives.