ABSTRACT
Creutzfeldt–Jakob disease (CJD) is a rare and fatal neurodegenerative disorder with a broad spectrum of early clinical manifestations, comprising neurological and psychiatric symptoms. The authors report the case of a patient admitted with a diagnosis of depressive disorder with psychotic symptoms, with post-mortem confirmation of CJD and discuss how CJD’s clinical heterogeneity can lead to misdiagnosis of the disease. Despite CJD’s unique pathogenesis, its kaleidoscopic presentation justifies the integrated investigation of patients with psychiatric symptoms, avoiding restrictive diagnosis.
LEARNING POINTS
- Creutzfeldt–Jakob disease is a rare and fatal neurodegenerative disease with a broad spectrum of early clinical manifestations, comprising neurological andpsychiatric symptoms.
- Because of its phenotypic heterogeneity and complexity and lack of more accurate pre-mortem diagnostic markers, misdiagnosis is easy.
- Despite CJD’s unique pathogenesis, its kaleidoscopic presentation justifies the integrated investigation of patients with psychiatric symptoms, even in the absence of additional typical neurological signs, avoiding restrictive diagnosis.
KEYWORDS
Creutzfeldt–Jakob disease, sporadic Creutzfeldt–Jakob disease, depressive disorder with psychotic symptoms
INTRODUCTION
Creutzfeldt–Jakob disease (CJD) is a rare neurodegenerative disorder characterized by the pathological deposition of the mutated prion protein (PrPCJD) in the central nervous system (CNS), causing neuronal loss[1]. The vast majority of CJD cases are sporadic (85%) (sCJD); the rest are to be considered familial, iatrogenic and variant forms[1,2].
Although it is classically defined by its neurological features (rapidly progressive dementia with myoclonus and other signs of CNS dysfunction), psychiatric symptoms may occur at the onset of sCJD[2,3]. No definite pre-mortem diagnostic test is available, but a diagnosis of sCJD is considered probable when any of the following criteria are met: distinctive symptoms associated with typical electroencephalogram (EEG) (periodic sharp wave complexes); magnetic resonance imaging (MRI) high signal abnormalities in the caudate nucleus and/or putamen on diffusion-weighted imaging or fluid attenuated inversion recovery; or a positive 14-3-3 cerebrospinal fluid (CSF) assay[2]. Death usually occurs within 1 year of symptom onset and definite diagnosis is made after PrPCJD neuropathological confirmation[2].
The authors describe a case of a patient admitted with a diagnosis of depressive disorder with psychotic symptoms, with post-mortem confirmation of sCJD, highlighting how sCJD clinical heterogeneity may lead to misdiagnosis.
CASE PRESENTATION
A 72-year-old man with a month-long history of fatigue, apathy, depressed mood, insomnia and confusion presented at the emergency department. He complained about mild hearing loss and gait imbalance for 3 months. The patient had hypertension and was under diuretic treatment; he lived alone, worked at a restaurant and had no previous history of substance abuse. Oropharyngeal congestion and bilateral tympanic depression were observed, with no focal deficits at neurological examination. Cranioencephalic computed tomography (CE-CT) displayed no alterations. The patient was diagnosed with peripheral vertiginous syndrome and depressive disorder and discharged with cinnarizine, betahistine, trimetazidine and sulpiride medication. In the following 3 weeks, the patient’s relatives noticed a tendency toward social isolation, irritability, emotional lability, difficulty concentrating, psychomotor retardation and visual hallucinations. At re-observation, the patient was agitated, presenting disorganized speech, memory impairment, gait imbalance, kinetic hand tremor and stereotyped movements of the limbs that stopped when he was distracted. Routine laboratory tests and new CE-CT were performed, revealing no alterations. The patient was admitted to the psychiatry department, with a possible diagnosis of depressive disorder with psychotic symptoms.
During the hospitalization, the patient exhibited rapidly progressive mental deterioration, alternating prostration and psychomotor agitation, culminating in a tonic–clonic seizure. In a few days, he became completely dependent, with urinary incontinence, need for nasogastric tube feeding and progressive akinetic mutism. Internal medicine and neurology collaboration was requested.
The post-critical EEG registered continuous slowing of brain activity (Fig. 1a) and a later EEG exhibited periodic sharp wave complexes (Fig. 1b).
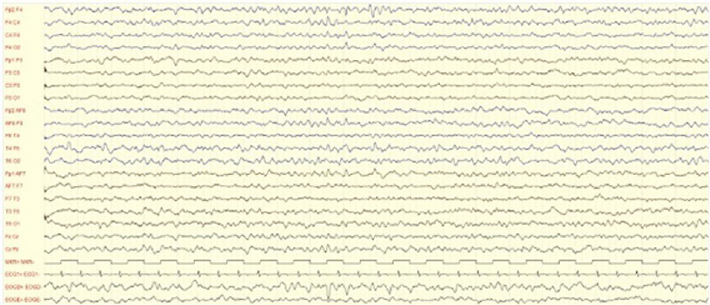
Figure 1a (click to enlarge)
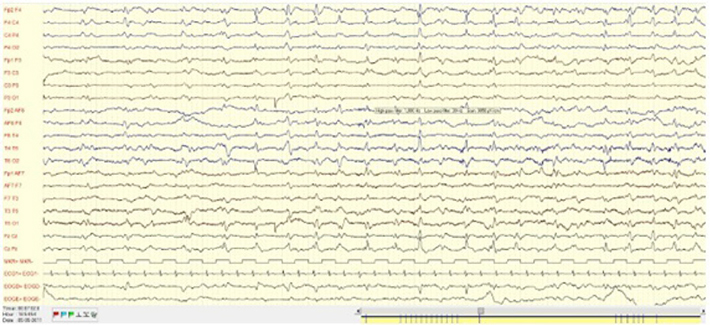
Figure 1b (click to enlarge)
Figure 1a: EEG continuous slowing of brain activity.
Figure 1b: EEG asynchronous periodic sharp wave complexes.
The MRI showed no specific alterations (Fig. 2).
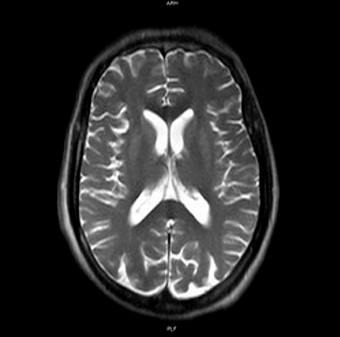
Figure 2a (click to enlarge)
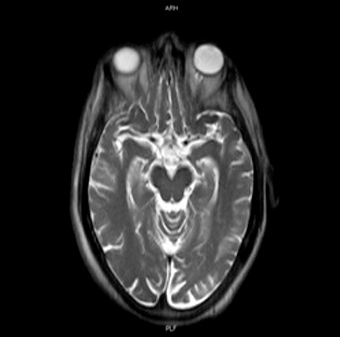
Figure 2b (click to enlarge)
Figure 2: Brain MRI: cortical atrophy, predominant in the parietal region, and normal basal ganglia
The diagnostic investigation revealed uncharacteristic results: neutrophilia without leukocytosis; no significant electrolyte imbalance; moderately increased serum immunoglobulin E, with normal complement, protein electrophoresis, thyroid and parathyroid hormones. Testing for viral and bacterial antibodies was negative (HIV and herpes virus, with immunity for cytomegalovirus; Treponema pallidum, Toxoplasma gondii, Borrelia burgdorferi, Coxiella burnetii, Leptospira species and Rose Bengal).
Blood and urine cultures were negative as serum tumour marker determination (CEA, CA 19.9, CA 125). CSF cytochemical analysis showed no alterations; culture, testing for Treponema pallidum, herpes virus, Cryptococcus, Mycobacterium and adenosine deaminase protein were negative; elevated β-amyloid, total tau and phosphorylated tau proteins and a clearly positive 14-3-3 protein titre were measured (Table 1).
| Protein | CSF concentration (pg/ml) | Reference values |
|---|---|---|
| β Amyloid | 663.6 | >500 pg/ml |
| Total tau | >12,000 | ≤ 60 years: 119.4±30.4 pg/ml > 60 years: 171.1±104.9 pg/ml |
| Phosphorylated tau | 89.2 | < 61 pg/ml |
| 14-3-3 | Strong positivity | Qualitative evaluation |
Table 1: Dementia biomarkers in CSF
The patient was empirically treated with ceftriaxone (2 g q12h), intravenous acyclovir (750 mg q8h) and dexamethasone (5 mg q8h). He died 27 days after hospitalization from nosocomial pneumonia and septic shock. A post-mortem neuropathological examination of the brain confirmed the diagnosis of sCJD, showing neuropil vacuolation and neuronal loss, with positive immunohistochemical staining for PrPCJD (Fig. 3a, b).
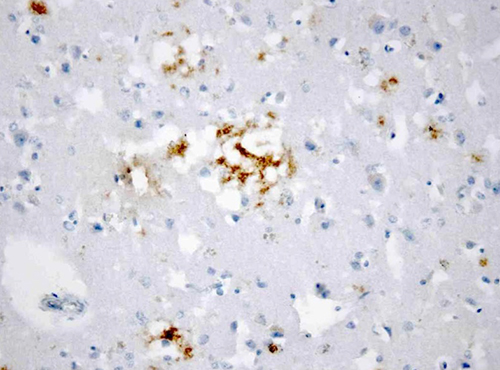
Figure 3a (click to enlarge)
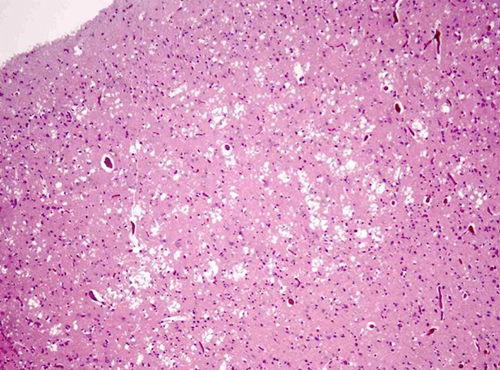
Figure 3b (click to enlarge)
Figure 3A: Post-mortem neuropathological examination of the brain. Neuropil vacuolation and neuronal loss (HE, 100×)
Figure 3B: Post-mortem neuropathological examination of the brain. Granular and perivacuolar PrPCJD deposits (immunohistochemical staining for PrP, 400×).
DISCUSSION
Twenty-six percent of patients with sCJD have psychiatric symptoms at disease onset, increasing to 80% within the first 100 days[3]. The patient presented with depression and behaviour and perception alterations as initial manifestations. Neurological signs, such as hyperkinetic movements and gait imbalance, were first interpreted as eventual adverse effects of the prescribed medication; in the literature, movement disorders induced by antipsychotics, such as sulpiride, and calcium channel blockers, such as cinnarizine, are reported[4–6]. The wide range of early psychiatric symptoms, the misinterpretation of the neurological signs as iatrogenic events and the lack of specific alterations in the first diagnostic approach would have justified the initial diagnosis of a depressive disorder with psychotic symptoms. Previous literature reports rare cases of sCJD patients presenting with depressive symptoms and psychosis, reinforcing the idea that sCJD phenotypic heterogeneity and complexity may delay and hamper its diagnosis[7,8].
Neurological dysfunction became prominent over the course of hospitalization, with rapid cognitive impairment, physical dependence and mutism. The initial blood analysis and CE-CT were important to exclude other diagnoses such as infections, malignancy, CNS space-occupying lesions, cerebral infarction and metabolic encephalopathies. The first EEG and the brain MRI did not present features typical of a diagnosis of sCJD, with periodic, though asynchronous, sharp wave complexes only seen in the later EEG. Notwithstanding the 97% diagnostic sensitivity for CJD of the 14-3-3 protein elevated titre in the CSF, the diagnostic specificity (78–92%) is lower, since this protein is an indicator of neuronal destruction, which may occur in other severe neurological conditions[9, 10]. Additional CJD pre-mortem diagnostic markers are needed. The high total tau protein titre and the reduced phospho-tau/total-tau ratio in CSF is emphasized and in line with the growing literature regarding the use of these parameters as diagnostic tools for CJD[11].
As psychiatric symptoms may occur in the early stages of sCJD, every neurological sign needs to be integrated within a holistic perspective. Despite CJD’s unique pathogenesis and rarity, its kaleidoscopic presentation demands a global perception of the patient, avoiding restrictive diagnosis.