ABSTRACT
Cutaneous angiosarcoma is a rare, highly malignant tumour of vascular endothelial origin. It usually arises in the skin and superficial soft tissue, mostly on the head and neck. It presents as a variety of lesions, and so is considered a great mimicker, leading to a delay in diagnosis and evidencing the importance of biopsy with immunohistochemistry confirmation. There are few reports of extremity involvement in patients with pre-existing chronic lymphoedema, or exposure to radiation therapy. We report the case of an 82-year-old woman with lower limb extensive cutaneous involvement, distant metastatic disease, and poor therapy response. Its rare location without predisposing factors highlights the need to raise awareness about this disease.
LEARNING POINTS
- Extremity involvement of cutaneous angiosarcoma has been rarely described. The marked heterogeneity in presentation leads to a delay in diagnosis and poor prognosis, so the index of suspicion should be high.
- The cases reported in the literature describe a well-known relationship between cutaneous angiosarcoma and predisposing factors, but its absence should not exclude the diagnosis.
- This case highlights the importance of recognizing and biopsy suspected skin lesions for immunohistochemistry diagnostic confirmation.
KEYWORDS
Cutaneous angiosarcoma, lower limbs, predisposing factors, immunohistochemistry
CASE DESCRIPTION
An 82-year-old woman presented to the emergency room with a 2-month history of a rapidly growing purplish plaque on the right leg. The patient had not previously undergone surgery or radiotherapy on her legs. Furthermore, there was no associated asthenia, weight loss, fever or local pain. Her past medical history was significant for arterial hypertension, stage 2 chronic kidney disease, a parathyroid adenoma excised 9 years earlier, and Alzheimer's disease.
The skin lesions started as small papules at the right ankle that rapidly grew in size and extension. Over time the lesion became an ulcerated plaque with an expressive haematic fluid. The patient was admitted with normocytic normochromic anaemia (haemoglobin 8.5 g/dl) and prerenal kidney failure secondary to dehydration by extensive exudation of the lower limb. Over time, she required two blood transfusions.
On physical examination, a large plaque located on the anterior aspect of the right leg was observed (Fig. 1). This lesion was composed of multiple papules and nodules involving the leg, distal thigh and dorsum of the foot in a multifocal pattern (Fig. 2). The papules and nodules were firm and painless upon palpation, showed a red-purple hue and had variable dimensions up to 10 mm. The anterior and medial aspects of the plaque were ulcerated. There was associated painless swelling of the lower extremity and elevated temperature. The patient was afebrile, and apart from obesity, there were no other abnormalities at physical examination, including the absence of palpable lymph nodes or hepatosplenomegaly.
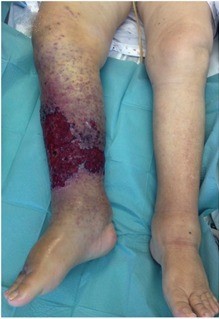
Figure 1 (click to enlarge)
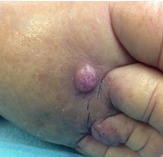
Figure 2 (click to enlarge)
Figure 1. Clinical features of extensive angiosarcoma and secondary oedema of the right lower limb
Figure 2 Purple, non-tender and dome-shaped tumour nodule
Methods and procedures
The blood cultures were negative. Doppler ultrasonography of the right lower extremity excluded deep venous thrombosis. An incisional biopsy on one of the nodules was performed, and the histopathological examination revealed a diffuse infiltrative neoplasia composed of large pleomorphic epithelioid cells infiltrating the deep dermis. There were areas with vascular-like channels lined with the same type of cells and several atypical mitotic figures were seen throughout (Fig. 3). Immunohistochemistry study disclosed expression of CD31, CD34 and factor VIII-related antigen, consistent with a vascular origin of the tumour cells (Fig. 4).
Computed tomography (CT) scanning of the chest, abdomen and pelvis revealed several enlarged pelvic, intra-abdominal lymph nodes and bone infiltration, suggestive of metastatic involvement. The CT scan of the lower limbs also identified the marked thickening of the soft tissues of the right leg infiltrating the muscular compartments of the extremity. Peripheral blood immunophenotyping, biochemistry and serological assessments, including β2-microglobulin, were normal, as were lactate dehydrogenase activity levels, while human immunodeficiency and hepatitis virus were negative” Lymph node biopsy was proposed but the patient and her family declined. Altogether, these findings supported the diagnosis of a cutaneous angiosarcoma, stage cT2bNxM1.
Considering the patient’s general condition, surgical excision to achieve local haemorrhage control was proposed but the patient and her family declined. She was medicated with a weekly palliative chemotherapy regimen with paclitaxel 100 mg/m2. After a total of eight chemotherapy cycles, a partial regression of the tumour was evident (Fig. 5). She died 4 months after diagnosis.
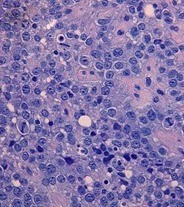
Figure 3 (click to enlarge)
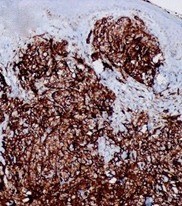
Figure 4 (click to enlarge)
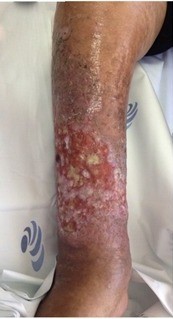
Figure 5 (click to enlarge)
Figure 3. The biopsy specimen disclosed a neoplasia with infiltrative growth in the dermis and hypodermis; the tumour consisted of epithelioid cells with an atypical nucleus and distinct nucleolus, sometimes displaying irregular poorly defined vascular spaces; frequent mitotic figures were present (H&E ×400)
Figure 4. Immunohistochemical expression of CD31 in tumour cells
Figure 5. Clinical aspect of the right leg 2 months after the initiation of chemotherapy, showing partial regression
DISCUSSION
Soft tissue sarcomas are a heterogeneous group that represents 1% of solid malignant tumours [1]. Angiosarcomas represent 1–2% of all sarcomas [2, 3] and can affect the skin, soft tissue, breasts or liver. The most common angiosarcoma site is the skin [2, 3]. Cutaneous angiosarcomas are extremely rare soft tissue tumours, of capillary and lymphatic endothelial cell origin. It presents more commonly in men with a median age of 73 years, mostly in the head and neck [3].
It is classified into three groups: (a) primary or idiopathic, the classic ‘head and neck-type’ (Wilson-Jones angiosarcoma); (b) secondary to ionizing radiation (radiation-associated angiosarcoma); and (c) secondary to chronic lymphoedema (Stewart-Treves syndrome) [2,3 4]. Our case is one of the few reported cutaneous angiosarcomas arising on the lower extremities in the absence of predisposing factors such as chronic lymphoedema or prior radiation therapy.
Clinically, cutaneous angiosarcoma usually resembles a bruise, blue to red colour, with a multifocal nodular component that develops as the tumour progresses [2,3]. Also, with increasing size, tissue infiltration, oedema, ulceration and bleeding can occur, as in our patient. This variety of lesions at initial presentation leads to a delay in diagnosis. Cutaneous angiosarcoma is considered to be a great mimicker, mimicking local bruises, eczema, deep infections, Kaposi's sarcoma, cutaneous metastasis, malignant melanoma, and lymphoma.
An adequate histological assessment is necessary and it is usually supported by immunohistochemistry features to establish the diagnosis[2]. Our case showed characteristic histopathological features, namely atypical endothelial proliferation of epithelioid and spindle cells, with frequent mitotic figures and necrosis[3]. Immunochemistry was the key to the diagnosis by identifying expression of CD31, CD34, and factor VIII antigen. CD31 is the most sensitive and specific marker and is present in 50% of cases.
Treatment of cutaneous angiosarcoma is challenging and optimal management has not been defined yet because of its rarity [2,4,5]. The gold standard treatment is complete surgical resection with wide margins and preoperative or postoperative radiation, due to the high propensity for local infiltration [3, 4]. Nonetheless, cutaneous angiosarcoma is often multifocal and permeates tissue far more widely than is clinically apparent, making complete surgical excision difficult to achieve; even with negative margin resection, approximately one-quarter to half of patients may face local recurrence. If tumours are unresectable or there is evidence of metastasis, palliative chemotherapy should be considered [2, 3], as in our case. Some treatments with chemotherapy are approved, such as taxanes [3,5]; it is noteworthy that cutaneous angiosarcoma is considered only modestly responsive to cytotoxic chemotherapy, as documented in this patient. New therapies are emerging, due to whole-genome sequencing. Reports on the use of antiangiogenic therapies such as VEGF-A monoclonal antibodies[3,4] and tyrosine-kinase[3] seem promising. Cutaneous angiosarcoma is an aggressive tumour with a high propensity for local recurrence and distant metastasis, most commonly by hematogenous spread to the lungs, liver, spleen and lymph nodes. The prognosis is very poor, with an estimated 5-year survival rate of 10–50% and a median overall survival rate of 6–16 months[3]. The worst prognostic factors include older age, tumour size larger than 5 cm, incomplete resection of the tumour, and distant metastasis [3,5]. In our case, the old age, the later diagnosis with local extension of the tumour and the distant metastasis in bone and lymph nodes (suggested by CT) were related to the poor prognosis.
In conclusion, we report a rare case of a cutaneous angiosarcoma because of its rarity and uncommon presentation in a woman with lower limb involvement without predisposing factors. This case emphasizes the clinical diversity of cutaneous angiosarcoma which may hinder diagnosis and the pivotal role of the biopsy and immunohistochemistry performed in time for any unsolved skin lesion to ensure accurate and timely recognition of this aggressive tumour.