ABSTRACT
Monoclonal gammopathies due to plasma cell dyscrasias can cause various rare neuromuscular disorders. The peripheral nervous system is most commonly affected, while muscle diseases associated with monoclonal gammopathies are rare. Skeletal myopathy, as a manifestation in the context of multiple myeloma, is extremely uncommon and is usually the result of immunoglobulin light chain (AL) amyloidosis deposits in the muscles. Here we present an atypical case of a patient with generalized myopathy as the presenting manifestation of light chain multiple myeloma. Interestingly, muscle involvement in our case was not the consequence of AL amyloidosis deposits but rather due to non-amyloid light chain deposition disease associated with light chain multiple myeloma.
LEARNING POINTS
- Light chain multiple myeloma can present as myopathy.
- Creatine kinase and muscle biopsy specimens only examined using routine stains can be normal in this condition.
KEYWORDS
Light chain deposition disease, skeletal myopathy, light chain multiple myeloma, monoclonal immunoglobulin deposition disease, chemotherapy
CASE DESCRIPTION
A 48-old-woman without a significant medical history, presented with 2 years of progressive and painless weakness of the proximal limbs. She had problems climbing stairs and difficulty carrying heavy objects. During the year before hospitalization, she also complained of dysaesthesia of the distal lower limbs, hoarseness of voice, low back pain and dyspnoea (New York Heart Association (NYHA) class III). A weight loss (15 kg) was also reported.
Physical examination showed a blood pressure of 130/65 mmHg and heart rate of 70 bpm, with normal respiratory and cardiac auscultation. Thickening of the skin of the face, eyelids, nose and lips in addition to macroglossia was observed (Fig. 1). Moderate facial weakness was noted.
Neurological examination revealed weakness of neck flexors (4/5), neck extensors (4/5), proximal muscles of the upper limbs (UL) (3–/5) and proximal muscles of the lower limbs (LL) (2+/5) scored according to the Medical Research Council (MRC) scale. Hypoesthesia in the distal LL was present. Deep tendon reflexes were absent in the LL and preserved in the UL.
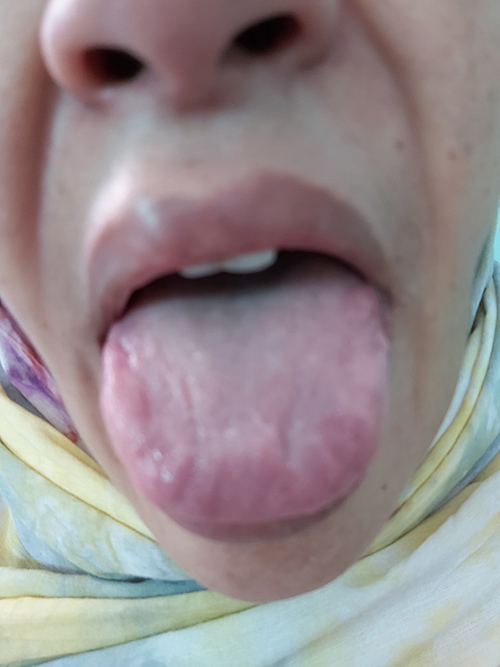
Figure 1 (click to enlarge)
Figure 1. Photograph revealing evidence of macroglossia
Methods and Procedures
Creatine kinase (CK) was normal at 105 IU/l (normal range: 24–170 IU/l) and nerve conduction study (NCS) revealed a myopathic pattern in the proximal muscles in addition to sensory and motor, axonal and demyelinating peripheral neuropathy in the LL with bilateral carpal tunnel syndrome. A muscle biopsy specimen (from the left tensor fascia lata (TFL), 2/5 MRC score) stained with haematoxylin and eosin (H&E), modified Gomori trichrome, nicotinamide adenine dehydrogenase (NADH) and periodic acid–Schiff (PAS) was unremarkable (Fig. 2)
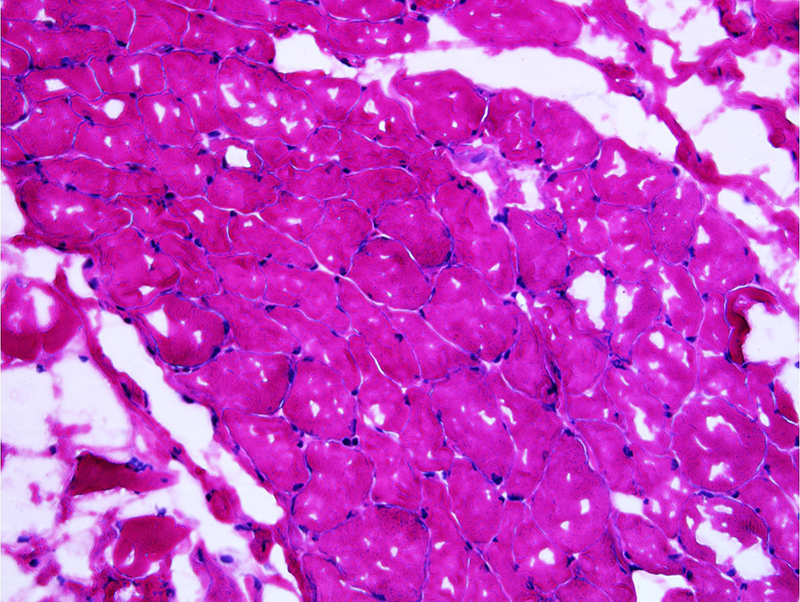
Figure 2 (click to enlarge)
Figure 2. Muscle biopsy showing regular muscle fibres and no remarkable lesions. Haematoxylin-eosin stain, magnification ×20
Acetylcholine receptor (AChR), antinuclear, anti-Ro(SSA), anti-La(SSB), anti-Sm, anti-U1RNP, anti-JO1, Scl70 and centromere antibodies were negative. Also, antiganglioside antibodies were negative. HIV and hepatitis B and C serology findings were unremarkable. TSH (thyroid-stimulating hormone) was slightly elevated at 7 mIU/l (normal range: 0.5–5.0 mIU/l) and fT4 (free thyroxine) was in the normal range. Anti-thyroid antibodies were negative.
An electrocardiogram (EKG) and chest X-ray were normal. The echocardiogram revealed hypertrophic cardiomyopathy (HCM). Cardiac magnetic resonance imaging (MRI) showed increased thickness of the cardiac walls and a late global subendocardial enhancement after gadolinium contrast injection. The left ventricular ejection fraction was decreased at 21.67%. These findings were highly suggestive of an infiltrative myocardial disease, particularly amyloidosis. Thus, the following investigations were carried out: 24-hour urine protein, which was positive at 2.6 g/24 h (normal range <0.15 g/24 h) and serum protein electrophoresis (SPEP) which demonstrated a spike in the gamma-globulin zone indicating a monoclonal gammopathy with hypogammaglobulinaemia. The monoclonal peak was estimated at 2.5 g/l. Serum immunofixation revealed two monoclonal bands corresponding to isolated free light chains of the lambda type. Lambda-type Bence-Jones protein was found and serum free light chains (SFLC) demonstrated highly increased free lambda chains at 6303 mg/l (normal range: 5.71–26.3 mg/l) and normal free kappa chains at 6.54 mg/l (normal range: 3.30–19.40 mg/l). The kappa/lambda ratio was 0.001 (normal range: 0.26–1.65 without renal insufficiency). These results confirmed the presence of a large amount of circulating monoclonal lambda light chains.
Bone marrow biopsy showed an excess of CD138 positive plasma cells at 30% which were monotypic lambda restricted. No congophilic deposits were observed in this biopsy specimen.
A body CT scan revealed ascites and pleural effusion, but no lytic bone lesions were observed. Due to her claustrophobia, the patient refused to undergo another MRI scan in order to look for bone lesions undetected by the CT scan. The work-up was also notable for the absence of anaemia, hypercalcaemia and renal insufficiency.
A diagnosis of lambda light chain multiple myeloma (LCMM) was made according to the 2014 updated International Myeloma Working Group (IMWG) criteria for the diagnosis of multiple myeloma (MM).
In view of the systemic involvement associated with LCMM, a search for AL amyloidosis was performed. A salivary gland biopsy with Congo red staining was negative. Also, supplementary Congo red staining of the previous muscle biopsy (left TFL) did not reveal any congophilic deposits. A sural nerve biopsy was indicated in order to identify amyloidosis but the patient refused it (and also refused renal biopsy). Taking into consideration this negative search for amyloidosis, non-amyloid light chain deposition disease (LCDD) as a diagnostic possibility was considered.
Thus, we performed immunohistochemistry (IHC) of the muscle biopsy specimen of the left TFL. This study demonstrated diffuse and intense reactivity with anti-lambda antibody in the endomysium, the perimysium and the vascular walls (Fig. 3). Thus, non-congophilic lambda light chain deposition in skeletal muscle was confirmed. A diagnosis of systemic LCDD associated with LCMM was finally made.
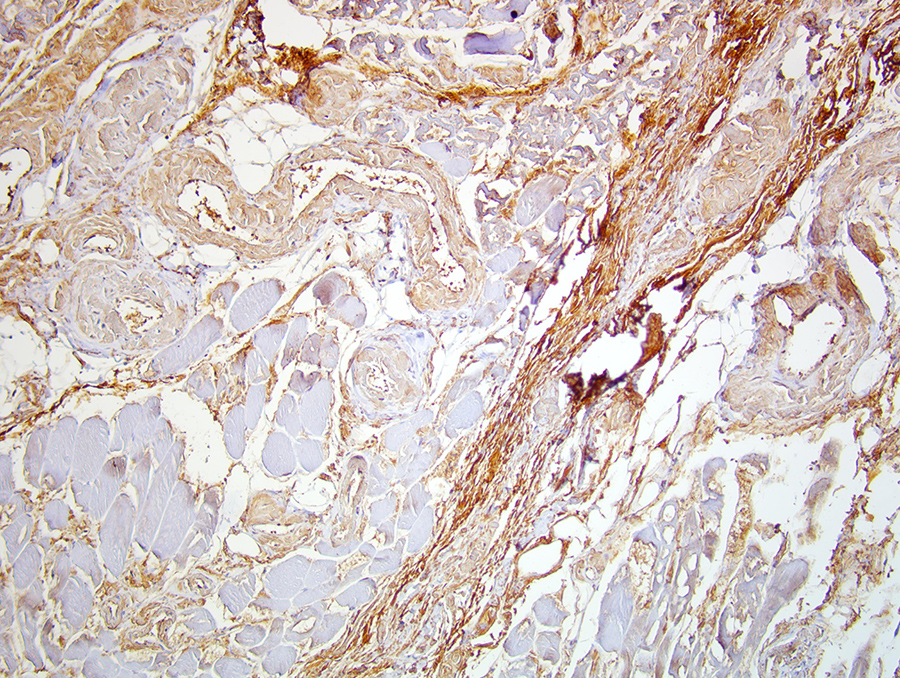
Figure 3 (click to enlarge)
Figure 3. Muscle biopsy revealing diffuse deposits of anti-lambda antibody. Immunohistochemistry, anti-lambda (Agilent/Dako polyclonal antibody), magnification ×20
The patient was started on a protocol of cyclophosphamide, bortezomib and dexamethasone (CyBorD). Pharmacological management of heart failure was initiated in addition to a low dose of levothyroxine for subclinical hypothyroidism.
DISCUSSION
LCMM is the third most common type of MM and has a poor prognosis. Its common manifestations include lytic bone lesions, anaemia, hypercalcaemia, renal failure, and an increased risk of infections. Extramedullary disease (EMD) mostly occurs later during LCMM progression. Only 1–2% of patients have EMD at the time of diagnosis and more than 90% of cases of EMD are diagnosed in the head and neck [1].
Skeletal myopathy, as a manifestation of EMD in the setting of MM, is very uncommon and is usually the consequence of AL amyloidosis deposition in the muscles [2, 3].
Our case has several distinguishing features. First, myopathy was the revealing manifestation of LCMM. Second, common manifestations related to LCMM such as anaemia, renal failure, hypercalcaemia and bone lesions (at least on CT scanning) were absent at the time of diagnosis. Third, skeletal myopathy in this setting of MM, was not the consequence of AL amyloidosis deposition but rather due to LCDD.
LCDD is a category of monoclonal immunoglobulin deposition disease (MIDD spectrum) and is characterized by tissue deposition of non-amyloid monoclonal immunoglobulin light chains in granular form, which are not stained by Congo red. These light chains are primarily deposited in the kidneys; however, the liver, heart, spleen, small bowel, skin and nervous system may also be affected. Approximately 11–65% of LCDD cases are associated with MM [4].
Interestingly, skeletal myopathy related to non-amyloid LCDD in the context of MM is extremely rare. In our case, the lambda light chain deposits were located in the endomysium along sarcolemmas, in vessel walls and in the perimysium. These locations are typical of MIDD as deposition most often occurs along the basement membranes of targeted tissues, suggesting that the monoclonal proteins likely have an affinity for some membrane component [5].
Like our patient, exceptional cases in the literature have reported monoclonal light chain deposits around muscle fibres that are negative on Congo red staining [6, 7]. But contrary to these cases, in which the deposits were kappa chain, our patient had lambda chain deposition in muscle, which represents another original feature of our observation. Indeed in LCDD, the light chain are generally of the kappa type while lambda light chains are rare [7].
In addition to skeletal myopathy, our patient also developed several characteristic manifestations related to LCDD such as nephropathy, heart failure with anasarca, skin lesions, macroglossia, laryngeal involvement, carpal tunnel syndrome and peripheral neuropathy. Subclinical primary hypothyroidism, in the absence of anti-thyroid antibodies and iatrogenic aetiologies, is likely the consequence of light chain deposition in the thyroid gland.
Our patient is receiving bortezomib, cyclophosphamide and dexamethasone. This combination therapy in MIDD is associated with high clonal response rates and prolonged progression-free survival [8].
Finally, our case demonstrates that MM can be revealed through very rare EMD manifestations such as myopathy. LCDD skeletal myopathy should be suspected in patients with muscle weakness who have evidence of MM, especially when muscle biopsy is negative for amyloidosis. Therefore, IHC is crucial in such circumstances for establishing the diagnosis of LCDD in the skeletal muscle.