ABSTRACT
Background: Peripheral nerve sheath tumors are benign or malignant. Schwannoma is a benign peripheral nerve sheath tumor originating from the Schwann cells that slowly grows eccentric to the nerve axis. Malignant transformation of a schwannoma is rare.
Case presentation: A 73 years old women that presented in our medical service for other medical problems was diagnosed with a tumor of the lower leg (a small mass neglected by the patient for about 10 years). Considering the ultrasound features of the tumor, it was suspected to be a schwannoma. The tumor was resected and the histopathological assessment revealed a schwannoma with areas of malignant peripheral nerve sheath tumor transformation.
LEARNING POINTS
- Malignant transformation of the schwannomas is rare but should be considered given its poor prognosis.
- We should distinguish between “ancient schwannoma” and malignant transformation of schwannoma since treatment and prognosis vary.
- Imaging is not entirely reliable in differentiating benign from malignant peripheral nerve tumors.
KEYWORDS
Schwannoma, malignant transformation, MPNST, malignant peripheral nerve sheath tumor.
INTRODUCTION
Peripheral nerve sheath tumours originate from nerve sheath cells and are classified as benign or malignant according to World Health Organization (WHO) criteria. The benign tumours of peripheral nerves include schwannoma, neurofibroma and perineurioma, the first two being the most common[1-3]. Malignant transformation of schwannomas is extremely rare[4].
Malignant peripheral nerve tumours are commonly referred to as malignant peripheral nerve sheath tumours (MPNST) and are primary malignant sarcomas originating from peripheral nerves or extraneural soft tissues that show nerve sheath cell differentiation[1,3,4].
CASE REPORT
We report the case of a 73-year-old woman who presented at the emergency department with high blood pressure (200/120 mmHg) and mild inspiratory dyspnoea. After blood pressure treatment and remission of symptoms, the patient was referred to our internal medicine department for further care and monitoring. The patient had also experienced chronic pain in the left ankle for approximately 10 years that had increased in intensity with associated loss of appetite and asthenia over the previous 6 months.
The patient’s past medical history revealed third grade essential hypertension with a very high cardiovascular risk (since 1984), coronary artery disease and a previous inferior myocardial infarction with revascularization by PTCA (in 2000), and diabetes mellitus type 2 (no insulin needed) since 2007.
At the physical examination, we noted first degree obesity, stasis crackles at auscultation of both lungs, no murmurs at auscultation of the heart, blood pressure 160/110 mmHg, heart rate 55/min (under beta-blocker treatment), and no functional neurological deficits (motor or sensory). Inspection of the lower legs showed an erythematous and mildly swollen area on the anterior surface of the left ankle (Fig. 1) which was painful on palpation.
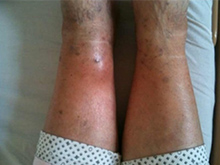
Figure 1A (click to enlarge)

Figure 1B (click to enlarge)
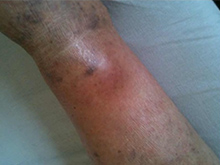
Figure 1C (click to enlarge)
Figure 1 : Physical examination of the left lower leg
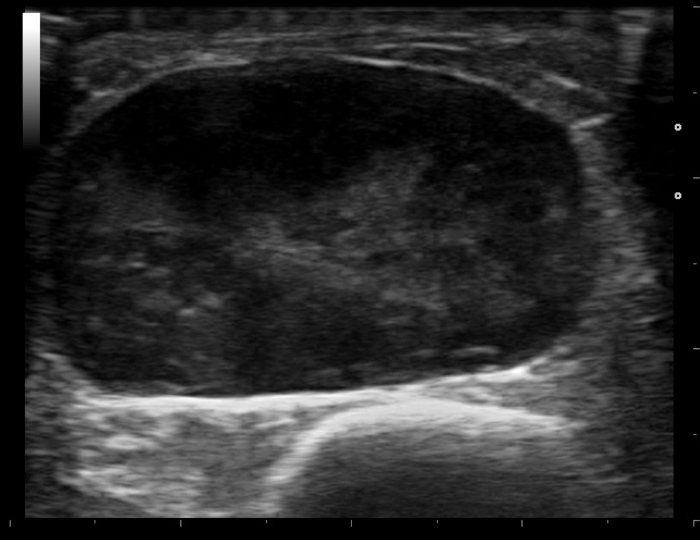
Figure 2 (click to enlarge)
Figure 2: Ultrasound image of the left lower leg mass.
Ankle ultrasonography was performed by an experienced operator (soft tissue ultrasonography being the most prompt and rapidly available investigation in our medical department) and a solid hypoechoic, well-delimited and well-vascularized (especially around the periphery) mass with posterior acoustic enhancement was seen (Fig. 2). The suspicion of a schwannoma was raised.
Given the pain in the lower leg, the weight loss and the patient’s wishes, it was decided to surgically resect the tumour. The patient was referred to our surgery department. Intraoperatively, the surgeon identified a firm, whitish mass about 3 cm in diameter, with increased consistency in contact with the saphenous nerve and including the origin of the great saphenous vein. Removal of the tumour also required removal of the origin of the great saphenous vein. The resected specimen was sent to the pathology department.
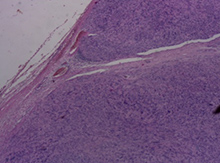
Histopathological assessment revealed, macroscopically, a resected mass measuring 3.6×3.2×2 cm with a bosselated surface. On cut section the mass was whitish-grey in colour with purplish and yellowish areas. Microscopic examination showed an encapsulated mass
partitioned by conjunctive tissue septa (Fig. 3a) and an area of geographic necrosis with palisading cells at the edges (Fig. 3b). Two types of cell populations were seen. One type consisted of hypercellular proliferation of spindle cells with a reduced quantity of cytoplasm and big, oval, vesicular nuclei with visible nucleoli, arranged in short intersecting fascicles (the common schwannoma component), while the other consisted of pleomorphic cells with large nuclei and frequent mitoses, arranged in solid sheets and isolated bizarre cells with giant, vesicular, hyperchromatic nuclei (the malignant MPNST component) (Fig. 3c). Histopathological assessment suggested schwannoma with areas of malignant transformation indicating low grade MPNST (Fig. 3).
There was no clinical suspicion of metastases. Chest, spine and pelvis x-rays were clear and abdominal ultrasound did not show any signs of secondary tumours. The final diagnosis was stage I MPNST of the left lower leg. The patient was referred to the oncology department, but refused further oncological treatment by declining to see the oncologist. She did not return for check-ups in our department, nor did she ever present to the oncology department requesting treatment.
1 (click to enlarge)

3 (click to enlarge)

3 (click to enlarge)
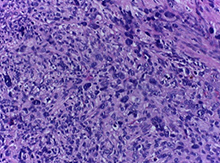
4 (click to enlarge)
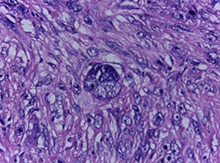
5 (click to enlarge)
Figure 3: Histopathology images of the tumor.
1 – Encapsulated mass with fibrous septa;
2 – Geographic necrosis area with palisading cells;
3 – Two cell populations;
4 – Malignant components at higher magnification 25x;
5 – A group of isolated bizarre cells at 40x magnification.
DISCUSSION
Schwannomas are benign tumours, most often solitary, that originate from Schwann cells. They represent approximately 5% of all benign soft tissue tumours and are the most common peripheral nerve tumours[4,5]. They usually grow slowly and eccentrically to the nerve fibre, and are thus encapsulated by the perineurium[4]. Most cases develop between 20 and 50 years of age, with equal sex distribution, the most affected sites being the head and neck and the flexor surface of the extremities[4,5]. Macroscopically, schwannomas are well circumscribed, round or oval whitish solid tumours, while microscopically they present a biphasic pattern with areas of highly organized spindle cells (Antoni A area) and less organized, hypocellular areas where myxoid tissue predominates (Antoni B area), surrounded by a true capsule. The originating nerve may be seen entrapped at the periphery of the tumour or along one border of the tumour. Immunohistochemistry shows expression of S-100 protein[3-5].
Malignant transformation of schwannomas is rare. The term ancient schwannoma, which is not an indicator of malignancy, is used to describe an old schwannoma that has undergone degenerative changes over time (this variant of schwannoma is also rare). Degenerative
changes that characterize an ancient schwannoma include interstitial hyalinization, cyst formation, calcificati on and haemorrhage, along with degenerative nuclear atypia, but without any mitotic activity[4,6,7].
MPNST are soft tissue neoplasms that have different types of differentiation towards one particular cellular structure of the nerve sheath. They account for 5–10% of all soft tissue sarcomas, have an incidence of 0.001% in the general population, and develop sporadically (40%), in association with neurofibromatosis type 1 (20–50%) or are radiation-induced (10%)[8-10]. MPNST arise either de novo or from a preexisting
benign precursor (most often from a neurofibroma and more rarely from a schwannoma). They are firm to hard tumours, tan-grey in colour with areas of necrosis or cystic transformation on cut section, with or without a fibrous pseudocapsule and showing invasion of the surrounding tissues. Histological diagnosis is usually challenging because of the pathological heterogeneity and absence of any specific findings at immunohistochemistry. Any sarcoma related to a major nerve having a nerve sheath line differentiation or clearly arising from a pre-existing benign nerve sheath tumour should be categorized as MPNST, unless proven otherwise. Microscopic features include a
‘marbled’ pattern of hypercellular fascicles of spindle cells interrupted by hypocellular myxoid areas, relatively large spindle cells with long, hyperchromatic, wavy or ‘serpentine’ nuclei, high mitotic activity, necrosis, and perivascular hypercellularity with indentation of cells into vascular lumens[3,8,10].
Magnetic resonance imaging (MRI) is the method of choice for the diagnosis of peripheral nerve tumours, although specific criteria for the differentiation of benign from malignant peripheral nerve tumours have not yet been established. Benign peripheral nerve tumours are seen as fusiform-shaped soft-tissue masses with low-to-intermediate signal intensity on T1-weighted images and high signal intensity on T2-weighted images that are contiguous with the nerve of origin, and show muscular fatty atrophy, complete split-fat sign and target sign. Larger (>5 cm), heterogeneous masses with central necrosis and ill-defined margins are more likely to be malignant[11].
The ultrasonographic features of benign and MPNST are variable and differentiation between schwannomas, neurofibromas and MPNST
is very difficult. Most often schwannomas appear as a solid, encapsulated mass along a peripheral nerve (eccentric to the nerve), which has
a homogeneous hypoechoic texture and posterior acoustic enhancement. Calcifications or cystic transformation may be seen Inside the
tumour[12,13].
Our patient had an undetected tumour of the lower leg for about 10 years that had slightly grown over time and become painful. Given
the histological appearance of the tumour with the features of a schwannoma that had some areas of MPNST and no features of ancient
schwannoma, we concluded it most likely was a schwannoma that underwent malignant transformation over the years.
The prognosis of MPNST is poor (20–60% of patients achieve 5-year survival), considering that they are aggressive, locally invasive tumours
and chemo-resistant. Association with neurofibromatosis type 1 is an independent indicator of poor prognosis. Other prognostic factors
are tumour size, local recurrence and complete surgical resection. Approximately 40% of MPNST are recurrent and two-thirds metastasize,
usually to the lung and bone[8,10,14-16].