ABSTRACT
22q11.2 deletion syndrome typically presents with congenital cardiac anomalies, immunodeficiencies and hypoparathyroidism. However, clinical findings vary greatly. We present the case of a 56-year-old man, with a history of cleft palate and schizophrenia, who was newly diagnosed with 22q11.2 deletion syndrome during an episode of hypocalcaemia. The syndrome is caused by developmental abnormalities of the embryonic pharyngeal arch system. Treatment of hypocalcaemia with oral calcium and vitamin D is usually sufficient.
LEARNING POINTS
- 22q11.2 deletion syndrome typically presents with congenital cardiac anomalies, immunodeficiencies and hypoparathyroidism.
- The syndrome has a variable phenotypic expression and can therefore remain unrecognised in adult patients with mild symptoms.
- 22q11.2 deletion syndrome should be borne in mind, particularly as regards adult psychiatric patients.
KEYWORDS
22q11.2 deletion syndrome, DiGeorge syndrome, hypocalcaemia, hypoparathyroidism
INTRODUCTION
22q11.2 deletion syndrome, also known as DiGeorge syndrome, is one of the most common microdeletion syndromes, with an estimated prevalence of 1 in 4000 live births [1]. The classic triad of clinical features are congenital cardiac anomalies, T-cell mediated immunodeficiency due to thymus hypoplasia, and hypocalcaemia caused by hypoparathyroidism [1–3]. Clinical findings in children include palatal defects, developmental delay, behavioural changes, facial dysmorphias, nasal speech, and renal and ophthalmic anomalies. Other clinical findings, particularly in adults, vary greatly, as does phenotypic expression [3, 4]. Due to the wide-ranging presentation of the syndrome in adulthood, the correct diagnosis may be difficult especially when the classic findings are mild or absent [1–4]. In this case report, we describe a patient who presented with 22q11.2 deletion syndrome in adulthood.
CASE DESCRIPTION
A 56-year-old Caucasian male was admitted to the Reinier de Graaf Hospital, Delft, The Netherlands, because of unconsciousness. He had been found by his brother lying on the ground at home. The patient had not answered his phone for several days before presentation. He was admitted to the intensive care unit (ICU) where he remained until his alertness improved. His remaining complaints were fatigue, malaise and muscle weakness, which he had also experienced before his acute illness. His medical history revealed a birth defect (palatoschisis), schizophrenia diagnosed at 35 years of age, cataract surgery at 54 years of age, and psoriasis. His GP medical chart disclosed one hypocalcaemic episode at the age of 45, which was treated with an oral calcium formulation without further investigation. Following a diagnosis of schizophrenia, the patient had lived in an assisted facility for patients with mental illness. An assigned mentor supported him in his financial affairs and assisted him in common social decisions. Prior to this episode, his close relatives had not been aware of his developmental abnormalities and intellectual disabilities. As far as we could determine, he had worked in IT, but his exact activities remained unclear to us. Finally, during the 6 months before his admission, he had lived completely independently, with the support of his mentor. Physical examination showed mild protrusion of the forehead, small lips, low-set ears and a hypoplastic mandible (Fig. 1), but no other abnormalities, especially not of the heart. Palatoschisis was noted by the speech therapist.
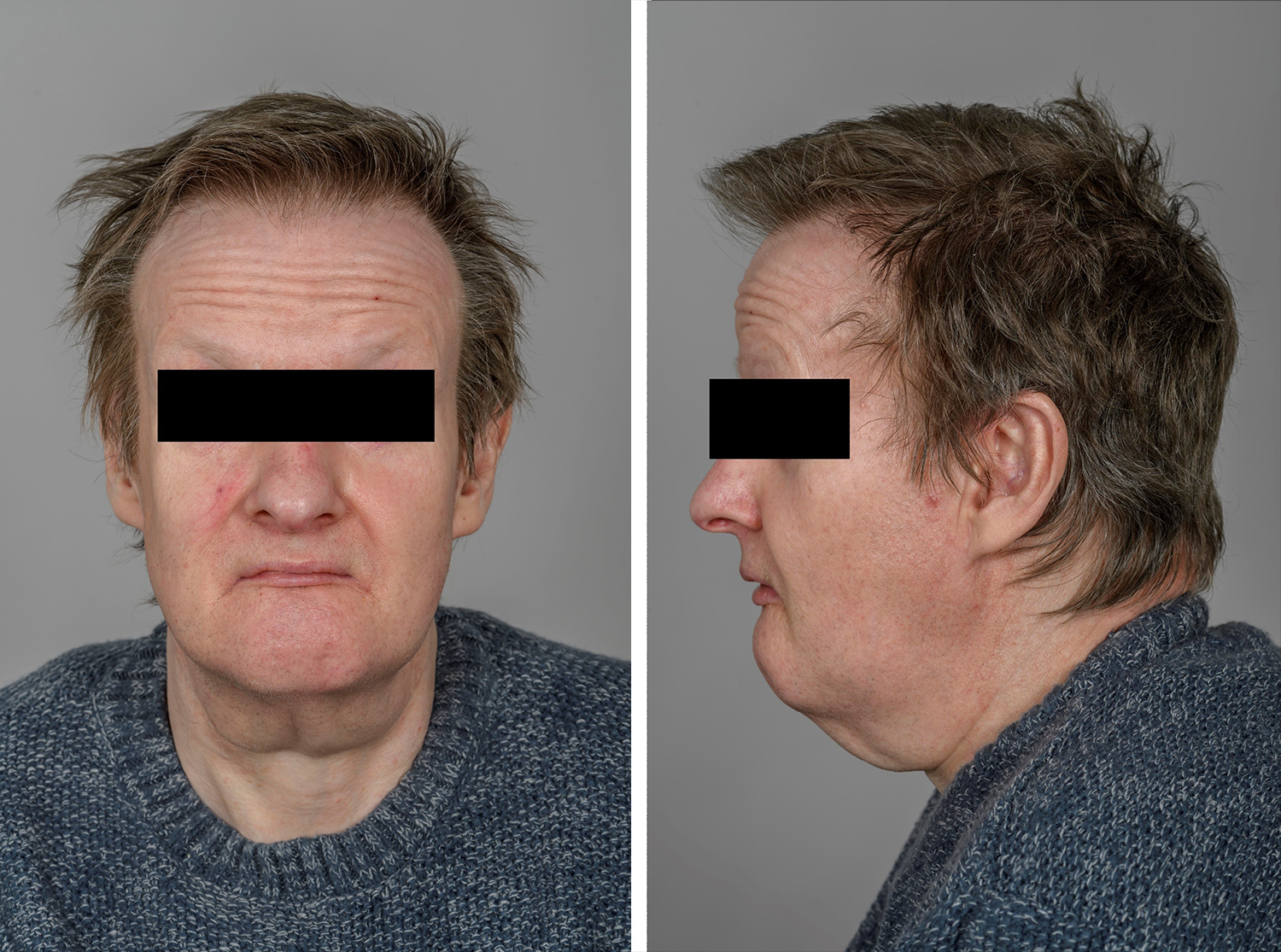
Figure 1 (click to enlarge)
Figure 1. Facial characteristics of the patient. Mild dysmorphic facial features include protrusion of the forehead, small lips, low-set ears and a hypoplastic mandible
Laboratory tests on ICU admission showed: C-reactive protein: 46 mg/l (reference value: 0–8 mg/l), haemoglobin: 10.0 mmol/l (8.0–10.5 mmol/l), leucocytes: 7.3×109/l (4.0–10.0×109/l), creatinine: 255 µmol/l (65–115 µmol/l), sodium: 179 mmol/l (135–145 mmol/l), magnesium: 0.86 mmol/l (0.65–1.00 mmol/l), calcium corrected for albumin: 1.04 mmol/l (2.15–2.55 mmol/l), phosphate: 4.17 mmol/l (0.70–1.45 mmol/l), 25-OH vitamin D: 21 nmol/l (50–250 nmol/l) and PTH: 0.75 pmol/l (1.6–8.2 pmol/l). Laboratory results from the ICU and from the general ward are shown in Table 1. Chest radiography disclosed an infiltrate in the upper lobe of the right lung. Electrocardiography showed a prolonged QTC interval of 620 ms but no other abnormalities. No abnormalities were found on sonography. The clinical symptoms led us to consider DiGeorge syndrome and genetic testing. The CytoScan HD Array (Affymetrix) confirmed a deletion on chromosome 22q11.2.
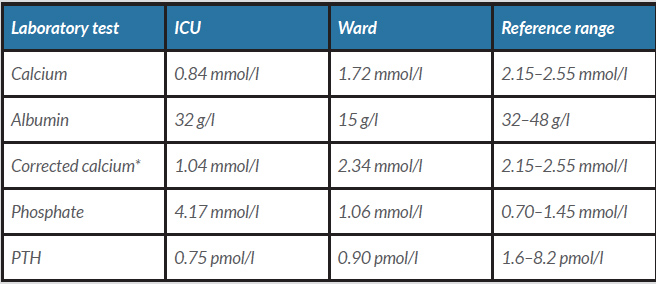
Table 1 (click to enlarge)
Table 1. Laboratory test results
* a Corrected calcium is calculated as serum calcium (mmol/l)+0.025×(40–albumin (g/l)).
Our patient was initially treated with intravenous fluids and antibiotics and received 2.0 g calcium gluconate and 3.0 g calcium carbonate daily. At discharge, he was being treated with calcium carbonate 1.0 g twice daily and alfacalcidol 0.5 µg once daily.
DISCUSSION
The phenotype of 22q11.2 deletion syndrome is variable [1, 3, 4] and in its classic presentation is usually diagnosed during childhood [2, 3]. In adolescence and adulthood, patients more often present with behavioural abnormalities, learning difficulties and psychiatric disorders. Hospital admissions are required in critical situations such as episodes of severe hypocalcaemia [3]. One series comparing children and adult 22q11.2 deletion syndrome patients showed a higher proportion of palatal abnormalities, mental disability and psychiatric illness among adults and a higher proportion of congenital cardiac abnormalities among children [5]. The most common findings in adults were minor facial anomalies [5], such as ocular hypertelorism, short palpebral fissures, hooded eyelids, low-set posteriorly rotated ears, a bulbous nose tip, micrognathia, microtia and a small mouth [1, 4, 6]. These facial dysmorphias have been described in other syndromes as well, but in combination with other clinical features can suggest 22q11.2 deletion syndrome [6]. The paediatric literature indicates that hypocalcaemia is caused by complete hypoparathyroidism caused by aplasia or hypoplasia of the parathyroid glands [7]. In contrast, parathyroid function is usually adequate in adults but may fail during episodes of increased metabolic demand, for example during stress caused by an infection or surgery [7].
The 22q11.2 deletion is a functional deletion and causes developmental abnormalities of the embryonic pharyngeal arch system, resulting in dysmorphogenetic and functional abnormalities of the thymus, thyroid, parathyroids, maxilla, mandible, aortic arch and cardiac performance due to changes in the outflow tract [3]. It is hypothesised that at least several of the 90 gene loci in the 22q11.2 region affect clinical expression [2, 3]. T-Box 1 (TBX1) is the best studied of these genes [3, 4]. Mutations of the TBX1 gene in mice models led to cardiovascular, thymic and parathyroid defects, although brain defects were lacking [3, 6]. Interestingly, several other loci within the 22q11.2 region are expressed in rodent central nervous system cells [3]. For example, the catechol-O-methyl transferase (COMT) gene located in the 22q11.2 region is involved in the enzymatic degradation of neurotransmitters such as dopamine and, to a lesser extent, serotonin [2, 3]. Mutation in this part of the genome results in impairment of neuronal maturation and is clinically associated with neuropsychiatric disorders such as schizophrenia, mood and anxiety disorders, and intellectual disabilities [2].
Recently, recombinant human parathyroid hormone (rhPTH(1-84)) was approved for use in hypoparathyroidism by US and European regulatory authorities. However, to our knowledge no treatment data are available in 22q11.2 deletion syndrome adults. Data on use of teriparatide (rhPTH(1-34)) in children, albeit limited, showed promising results. Subcutaneous injection of teriparatide 20 µg once daily improved hypocalcaemic symptoms in two children with 22q11.2 deletion syndrome and persistent hypocalcaemia despite treatment with daily oral calcium formulations and vitamin D. Teriparatide add-on also resulted in an increase in serum calcium and fewer hospital admissions during a mean follow-up of 2.5 years [8]. Whether teriparatide is non-inferior to the complete rhPTH(1-84) peptide in 22q11.2 deletion syndrome patients remains unknown.
In summary, this case report of a man presenting with 22q11.2 deletion syndrome highlights the variable phenotypic expression of this abnormality. The syndrome is rarely diagnosed in adult patients and frequently unrecognised in chronic patients, particularly those with psychiatric syndromes and mild dysmorphic features. 22q11.2 deletion syndrome should not be overlooked in adult psychiatric patients, particularly those with mild hypocalcaemia and/or hypocalcaemic episodes.