ABSTRACT
Mucormycosis is a rare fungal infection that often causes rhinocerebral disease. However, there have been rare cases of mediastinal involvement. These patients remain a therapeutic challenge and mortality in this group is very high. We report a case of mediastinal mucormycosis with invasion of the heart and right lung in a patient with chronic granulomatous disease (CGD) and also review the available literature on mediastinal mucormycosis.
LEARNING POINTS
- Mucormycosis is a very rare cause of mediastinal mass, and has a high risk of mortality.
- Early recognition and treatment will likely increase the patient’s chances of survival.
- Chronic granulomatous disease (CGD) is associated with an increased risk of fungal infections and should be considered for itraconazole prophylaxis.
KEYWORDS
Mucormycosis, chronic granulomatous disease, fungi
INTRODUCTION
Mucormycosis is a rare destructive fungal disease, commonly presenting as rhinocerebral disease in hyperglycaemic metabolic acidosis patients. Diagnosis of this fulminant aggressive disease is very important as prompt surgical control and swift medical management of infection reduces mortality and organ loss [1]. Diagnosis requires a pathological examination of the tissue involved as swabs and washing specimens are inadequate. We report a case of advanced invasive mediastinal mucormycosis in a patient with chronic granulomatous disease (CGD).
CASE DESCRIPTION
CASE DESCRIPTION
Our patient was a 21-year-old man who had been diagnosed with CGD 4 years previously and had a positive family history with one sister with CGD. He was kept on long-term sulfamethoxazole/trimethoprim prophylaxis therapy.
The patient presented to his local hospital with fever, shortness of breath, night sweats, cough and chest pain of 3 weeks’ duration. The initial diagnosis was a lung abscess as the chest radiograph showed a large right parahilar mass. Surgical resection was attempted but it was reported that the mass was surgically unresectable, and the patient was referred to our hospital for further management.
Upon presentation to our hospital, the patient was ill-looking with intermittent fevers, tachycardia, frequent premature atrial contractions and a few episodes of supraventricular tachycardia (SVT). Physical examination showed deviation of the trachea to the left, and decreased air entry over most of the right hemithorax with dilated neck veins and raised jugular venous pressure (JVP) suggestive of superior vena cava syndrome.
A chest radiograph was obtained and showed a large mass occupying the right hemithorax with mediastinal shift to the left side (Fig. 1). Computed tomography (CT) of the chest showed a large right hemithorax mass lesion measuring 11.2×10×12 cm with enhanced periphery invading the mediastinum, superior vena cava, right and left atria, part of the inferior vena cava, diaphragm, liver and anterior chest wall (Fig. 2). The mass was also causing compression of the heart and right pulmonary artery.
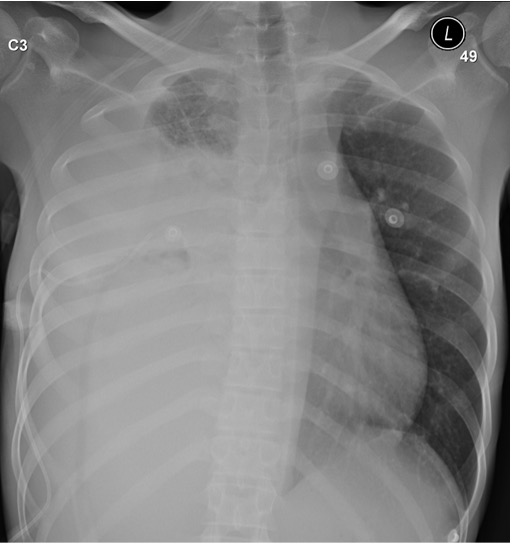
Figure 1 (click to enlarge)
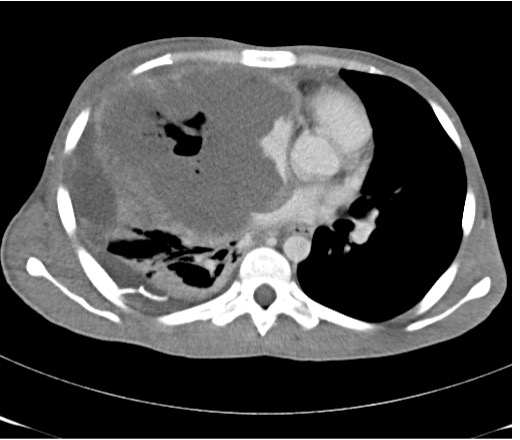
Figure 2 (click to enlarge)
Figure 1. Chest x-ray on admission showing opacification of almost all of the right hemithorax with shift of the mediastinum to the left
Figure 2. CT of the chest on admission showing a large mass with a necrotic centre with extensive involvement of mediastinal structures and the right side of the heart
Bronchoscopy with broncho-alveolar lavage was non-diagnostic. Positron emission tomography (PET) using fluorodeoxyglucose (FDG) was obtained and showed a large mass with central necrosis with increased uptake. Fungal culture from pleural body fluid was positive for Rhizopus species.
After consultation with thoracic surgery, it was decided to attempt exploration with maximum debulking. In the operating theatre a large mediastinal mass was noted to be invading the right atrium, right ventricle, right lung, chest wall, right mainstem bronchus as well as the inferior vena cava and the pulmonary vein close to the left atrium. During an attempt to remove it, the inferior vena cava was injured but repaired. At that point it was decided that the mass was inoperable and after control of bleeding, the chest was closed. Biopsy of the mass revealed necrotizing granulomatous inflammation with fungal organisms identified, with cultures being positive for Rhizopus species.
Due to failure of surgical debulking, the plan was for medical management only and the patient was started on double antifungal therapy, namely liposomal amphotericin and caspofungin. The patient remained ventilated and subsequently a tracheostomy tube was inserted. Over the following weeks, the patient’s general condition stabilized and episodes of supraventricular arrhythmias ceased. A follow-up chest radiograph and CT of the chest showed a decrease in the size of the mass (Figs. 3 and 4). However, 6 weeks after a second attempted surgical resection, the patient deteriorated rapid over a few hours with refractory shock and subsequently died.
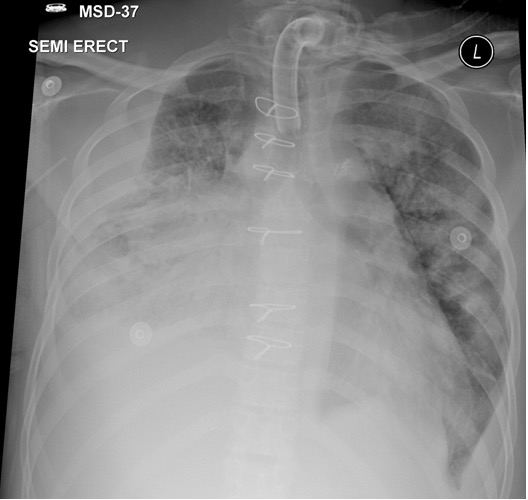
Figure 3 (click to enlarge)
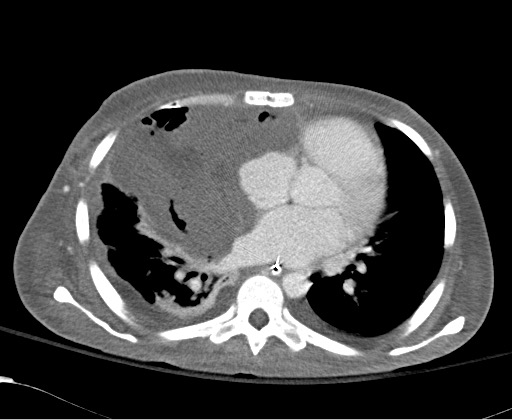
Figure 4 (click to enlarge)
Figure 3. Chest x-ray after 4 weeks of treatment showing improved aeration of the right lung
Figure 4. CT of the chest showing partial resolution of the mass after 5 weeks of treatment
DISCUSSION
Chronic granulomatous disease (CGD) is a rare inherited disorder that occurs with a prevalence of approximately 1/250,000 live births, and is characterized by a faulty NADPH oxidase system, culminating in an inability of phagocytes to generate superoxide, resulting in defective killing of pathogens [2]. This can lead to infections with Staphylococcus aureus, Pseudomonas species, Nocardia species, and fungi including yeasts and moulds. Aspergillus species, Candida, Cryptococcus and Histoplasma are the fungi most implicated in causing invasive infections in patients with CGD [3].
Dotis et al. [4] performed a literature review of all published non-Aspergillus fungal infections in CGD patients in 2013 and reported 68 cases in 65 CGD patients. The most prevalent non-Aspergillus fungal infections of CGD were caused by Rhizopus spp. and Trichosporon spp. found in nine cases each. The most commonly affected organs were the lungs alone or in combination with other organs in 69.9% of cases, followed by soft tissue infections in 17.6%.
The increased risk of infectious complications in CGD patients has led to the introduction of prophylactic strategies over the last two decades. Mouy et al. [5] first looked at the efficacy of long-term itraconazole prophylaxis over 6 years among 32 patients with CGD in preventing Aspergillus infections. They observed that daily administration of itraconazole was well tolerated, and significantly reduced the frequency of severe invasive Aspergillus infections among CGD patients when compared with historical controls. More recently, Beauté and colleagues [6] evaluated the impact of itraconazole prophylaxis among 155 CGD patients from 1976 to 2008 and observed a significant reduction in invasive fungal disease (IFD) in the group that received itraconazole prophylaxis when compared with the no prophylaxis group (0.027 versus 0.053 IFD/patient-years, p<0.01). These findings have led many to argue for routine anti-fungal prophylaxis in CGD patients. The fact that our patient was not receiving itraconazole prophylaxis placed him at increased risk of IFD and also points to the need to make physicians aware of the need for anti-fungal prophylaxis to prevent life-threatening IFDs.
In 2019, Cooper et al. [7] performed a literature review and identified 12 cases of mediastinal mucormycosis. The median age was 35 years and 11 of 12 patients had complications related to the mass, like our patient did. Reported complications included atrial fibrillation, complete heart block, acute paraplegia, superior vena cava syndrome, pericardial effusion, bronchial perforation and vocal cord paralysis. Unfortunately, only two of these 12 patients survived. Both of the survivors had parahilar lesions that did not cause complete collapse of the right lung and mass effect on the left, as our patient had. One of them, who was 22 months old, had a thoracotomy, followed by 15 weeks of amphotericin B treatment and then 12 weeks of itraconazole. The second patient was a 76-year-old woman who refused surgery but survived after receiving amphotericin B for 11 weeks.
We performed a literature review of the period between the review of Cooper et al. [7] and the present and identified three other cases. One was a patient who developed pneumopericardium and died [8], a second case was a patient with a 7.2×5.4 cm mediastinal mass who received isavuconazonium sulfate and micafungin followed by surgical resection and survived [9]. The last case was a patient who initially had pulmonary mucormycosis which invaded the airways and mediastinal structures, causing a bronchomediastinal fistula and eventual death due to haemorrhage from pulmonary vasculature invasion [10].
The addition of these three cases and our own increase the total number of mediastinal mucormycosis cases to 16, of whom three (18.75%) survived.
Based upon the available evidence, we believe that early recognition of the disease gives the best chance for cure. Investigation methods include imaging but more importantly tissue diagnosis. As regards pulmonary mucormycosis infection, there are reports of patients with early pulmonary infection who were cured with lobectomies [11]. Early lobectomy and surgical debulking remains the mainstay for successful treatment combined with prolonged systemic antifungal therapy. Reports of combination therapy especially with echinocandins have suggested improved outcome and should be considered when dealing with such patients [12]. However, when there is mediastinal involvement, our experience and that of others shows that although the overall principles of treatment remain the same, the prognosis in these patients is worse with a greater than 80% mortality rate.
CONCLUSIONS
Mucormycosis is a rare complication of CGD. Clinicians should have a high clinical suspicion to ensure timely tissue diagnosis. It should be in the differential diagnosis of cavitary lung lesions in addition to other fungi. Delayed diagnosis and/or late presentation of mucormycosis have a very poor prognosis with limited surgical options. Medical treatment is often unsuccessful.