ABSTRACT
The VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome is a recently described X-linked autoinflammatory condition caused by a somatic mutation of the UBA1 gene and characterized by an evolving phenotype. This includes inflammatory processes such as recurrent fever, Sweet’s syndrome of the skin, pulmonary fibrosis, relapsing polychondritis and venous thromboembolism. An important feature, present in almost all cases, is the development of a macrocytic anaemia with vacuolization of myeloid and erythroid precursors. Usually, these patients require high doses of steroids to control symptoms and respond poorly to disease-modifying drugs.
We describe a new case of the VEXAS syndrome presenting with Sweet’s syndrome which has now been followed for 6 years.
LEARNING POINTS
- An inflammatory syndrome with skin and pulmonary involvement in an elderly male patient with haematological abnormalities such as a macrocytic anaemia, myelodysplastic syndrome or venous thrombotic events should raise suspicion of the VEXAS syndrome.
- Close collaboration between rheumatologists and haematologists is important in diagnosing and managing this complex disorder.
KEYWORDS
Inflammatory syndromes, somatic mutation, macrocytic anemia
CASE DESCRIPTION
The patient was first seen in 2015 at the age of 72 years with constitutional symptoms, a recent deep venous thrombosis of the left leg and new cutaneous lesions (Fig. 1). His previous history included a myocardial infarction in 2012 and mild psoriasis. Laboratory evaluation was notable for a high ESR of 100 mm/h and elevated CRP (121 mg/l) with negative screening for vasculitis or connective tissue diseases. Blood cultures were negative, as were multiple serologies for viral or bacterial pathogens. Haemoglobin was 121 g/l and MCV 102 fl. A skin biopsy showed dense infiltration by granulocytes compatible with histiocytoid Sweet’s syndrome. Pulmonary infiltrates with an UIP pattern and some pleural effusion were found on a CT scan of the lungs. Broncho-alveolar lavage revealed an increased cell number (405×106/l) with slight granulocytosis of 6%, and lung function tests demonstrated a reduced diffusion capacity (DLCO 35%). No underlying malignancy was found by PET-CT and a bone marrow examination showed only minimal signs of dysplasia but no evidence of a haematological malignancy.
A presumptive diagnosis of Sweet’s syndrome with pulmonary fibrosis was made. The patient was treated with prednisolone at a dose of 1 mg/kg/day. He initially responded well but required doses above 20 mg/day. Several steroid-sparing therapies were tried (methotrexate, azathioprine and mycophenolate mofetil) without benefit.
Treatment with colchicine allowed prednisolone to be reduced to 7.5 mg/day with good control of fever and night sweats. Temporarily, the ESR normalized but the patient continued to lose weight (total of 25 kg in 5 years).
Because of increasing anorexia, progressive macrocytic anaemia, a rise in the ESR to more than 100 mm/h, a further decline in DLCO (28%) and recurrent deep venous thrombosis, a second diagnostic work-up was carried out in 2020. Again, no malignant process was found. PET-CT showed progressive pulmonary infiltrates, and bone marrow cytology minimal dysplastic changes. This time vacuolization of myeloid precursors was noted (Fig. 1). Serum levels of IL-6 were markedly elevated (79.8 pmol/ml, normal <7 pmol/ml), while those of TNF-alpha and IL-1 were normal. The therapy-refractory course and the vacuolization of bone marrow cells prompted genetic investigation for the recently described VEXAS syndrome. DNA was isolated from unselected bone marrow cells and subjected to sequencing of position 41 of the UBA1 gene using bidirectional primers (sequences on request). An A>G (p.41Met>Leu) mutation was found (Fig. 1), confirming the diagnosis. Treatment with tocilizumab was planned but could not yet be started due to recent infectious and cardiovascular complications.
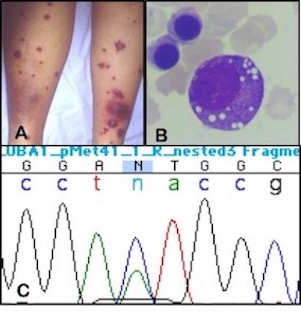
Figure 1 (click to enlarge)
Figure 1. (A) Clinical picture of the skin with tender erythematous plaques as seen at initial presentation. (B) Bone marrow aspirate showing extensive vacuolization of myeloid precursors. (C) Sanger sequencing of unpurified bone marrow cells detects a A>G mutation at position p.Met41 of the UBA1 gene, mosaicism is indicated by a normal sequence in a minority of cells
DISCUSSION
The recently described VEXAS syndrome is an exciting discovery [1, 2]. The identification of an acquired somatic mutation causing an adult onset autoinflammatory syndrome is of great interest to both rheumatologists and haematologists. Here we report a new patient with this syndrome. The clinical presentation and the results of the mutational analysis correspond very well with the original report. Our patient had a chronic progressive multisystem inflammatory syndrome with impressive constitutional symptoms, pulmonary fibrosis and neutrophilic dermatosis. A clue to the diagnosis was the finding of a macrocytic anaemia which is unusual in patients with inflammatory conditions, unless treated with drugs interfering with DNA synthesis. The anaemia of chronic inflammation usually is hypochromic and microcytic. Unexplained macrocytic anaemia in such patients should therefore raise suspicion of the VEXAS syndrome and referral to a haematologist is recommended. Bone marrow examination should be performed with careful examination to look for vacuoles in myeloid and erythroid precursors which are characteristic of the VEXAS syndrome. Ultimately, the diagnosis is confirmed by sequencing for a UBA1 gene mutation. So far, all mutations have involved amino acid p.41Met resulting in the loss of the canonical cytoplasmic isoform of UBA1.
In the 25 patients with the VEXAS syndrome described so far, a treatment-refractory course was typical. Ten patients died either as a result of their disease or treatment-related complications, indicating a bad prognosis. Our patient also required high doses of steroids and did not respond to several disease-modifying drugs. Only colchicine demonstrated some limited effects on symptoms and steroid requirement.
About 40% of patients in the original publication were treated with colchicine, but no specific responses were reported. High levels of serum IL-6 were also found in our patient and could be an additional characteristic feature. Given this observation, tocilizumab could be an interesting candidate for further evaluation. The effectiveness of tocilizumab in other conditions characterized by high serum IL-6 levels, such as adult-onset Still’s disease [3], might suggest a role also in treatment of the VEXAS syndrome.