ABSTRACT
Background: Neurofibromatosis type 1 (NF1), also known as von Recklinghausen disease, is a one of the more common hereditary autosomal disorders. However, osteomalacia in neurofibromatosis type 1 is very rare tumour-induced osteomalacia; fibroblast growth factor-23 is usually implicated.
Patients and methods: We report the case of a patient with a history of von Recklinghausen neurofibromatosis who presented with hypophosphataemic osteomalacia.
Results: The patient was treated with high-dose calcitriol and oral phosphate with clinical improvement.
Conclusion: Even though it is a rare entity, we must consider the diagnosis of hypophosphataemic osteomalacia in patients with neurofibromatosis in order to deliver appropriate treatment.
LEARNING POINTS
- Osteomalacia during von Recklinghausen disease is a rare presentation of an uncommon condition and has a poorly understood mechanism.
- The treatment of oncogenic osteomalacia includes tumour removal which, however, is not always possible.
- Administration of calcitriol alone is not sufficient and phosphorus intake is mandatory to improve symptoms.
KEYWORDS
Osteomalacia, neurofibromatosis, hypophosphatemia
INTRODUCTION
Neurofibromatosis type 1 (NF1), also known as von Recklinghausen disease, is one of the most common hereditary autosomal disorders, affecting approximately one person in every 3000–4000 [1]. NF1 is characterized by extreme variability in its clinical expression, even within the same family. The clinical presentation of NF1 includes specific features (neurofibromas, café-au-lait spots on the skin, axillary and inguinal lentigines, Lisch nodules) and tissue alterations (nervous, vascular). Osteomalacia presenting during von Recklinghausen disease is rare and has a poorly understood mechanism. Since the first description of this association in 1918 by Gould, fewer than 40 cases have been reported in the literature. Here we describe a case of neurofibromatosis which presented with clinical and radiological features of osteomalacia.
CASE DESCRIPTION
A 67-year-old man with a history of hypertension, presented to the rheumatology department of our institute with progressive bone pain and difficulty walking for several months. The patient was a known case of neurofibromatosis type 1 since early adulthood, which had presented initially with café-au-lait spots followed by multiple cutaneous neurofibromas. His two sisters were also affected. The patient began experiencing progressive bone pain affecting his thighs several months previously, forcing him to walk with the help of crutches. There was also diffuse joint pain, especially at the level of both shoulders.
Examination of this patient, who was otherwise in good general health, showed numerous plexiform neurofibromas of different sizes all over his face, neck, back and abdomen, as well as café-au-lait spots (Fig. 1), at least 20 of which measured over 20 mm in diameter. There was no asymmetry or deformity of the extremities. In addition, we noted limitation of the abduction and the internal rotation of both hips as well as muscular weakness of both lower limbs.
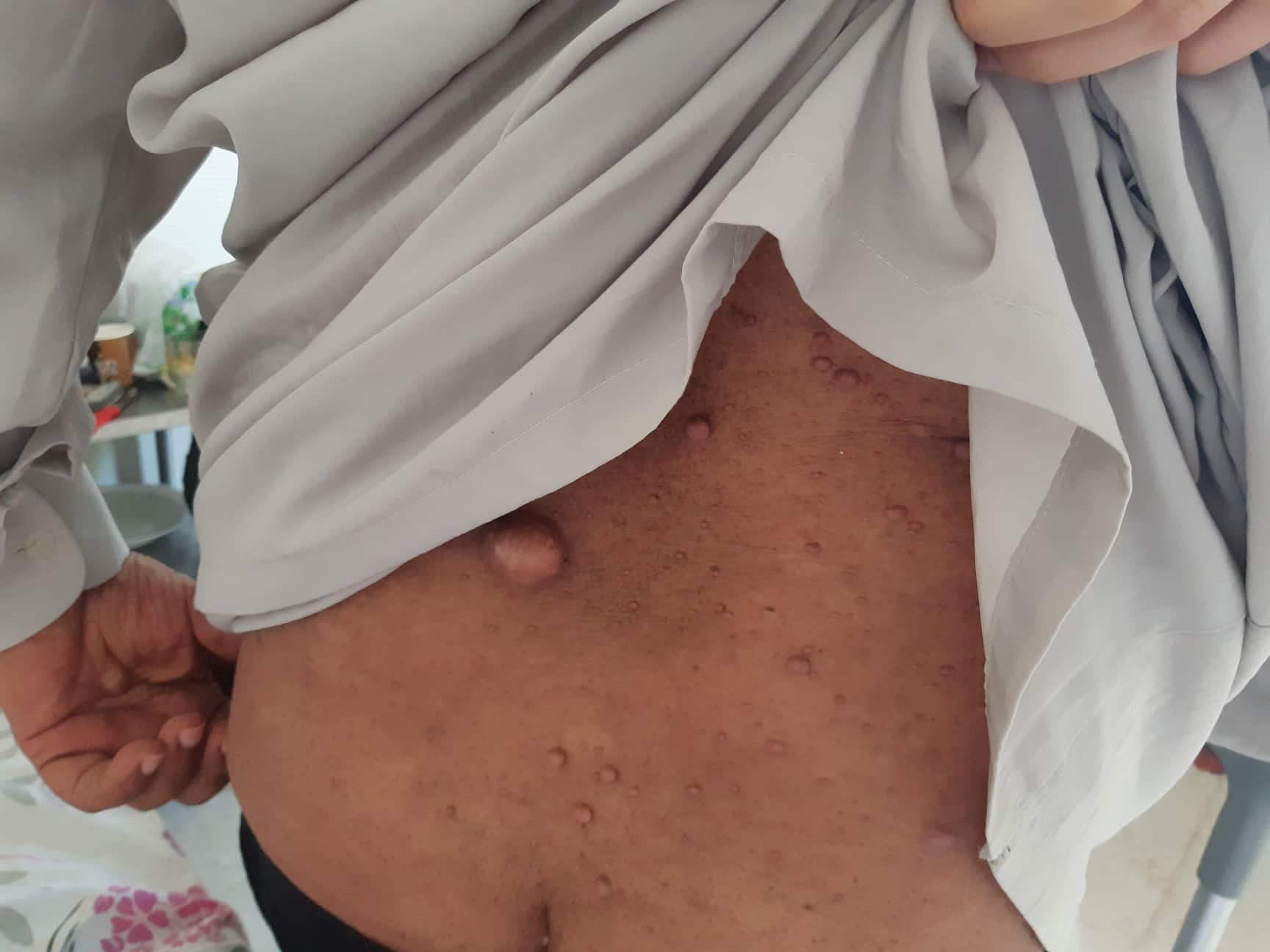
Figure 1 (click to enlarge)
Figure 1. Numerous plexiform neurofibromas on the back
Radiography of the pelvis with bilateral hips revealed a hyper-transparent fuzzy bone structure at the pelvis and Looser's zone fractures of the upper shafts of both femurs. A radiograph of the spine showed biconcave vertebrae. The radiological features were consistent with a diagnosis of osteomalacia. Laboratory data revealed normal serum calcium but low serum phosphorus (0.51 mmol/l). The 24-hour urinary excretion of calcium was low at 1.2 µmol/l, but phosphorus was high at 27.03 mmol/l. Alkaline phosphatase levels were twice normal, while serum parathyroid hormone (PTH) levels were six times normal. On the basis of the radiological and laboratory findings, a final diagnosis of hypophosphataemic osteomalacia in a patient with von Recklinghausen disease was made. Our patient is on regular follow-up. High-dose calcitriol and oral phosphate were prescribed, but surgical resection was not advised in view of the numerous neurofibromas. The patient’s constitutional symptoms have improved, but the fractures have shown no radiological signs of healing in the last 3 months of follow-up.
DISCUSSION
Osteomalacia is often caused by vitamin D deficiency, in which case hypophosphataemia is usually due to secondary hyperparathyroidism. However, in other forms of osteomalacia, the primary defect is hypophosphataemia, also known as hypophosphataemic osteomalacia. The association between hypophosphataemia, hyperphosphaturia and inadequate response to oral calcium and vitamin D therapy suggests renal, oncogenic or hereditary X-linked hypophosphatasia [2].
Oncogenic hypophosphataemic osteomalacia (OHO) is a rare endocrinological paraneoplastic syndrome but probably still the most common cause of acquired hypophosphataemic osteomalacia in adult males [3]. Patients range in age from 7 to 77 years with a male:female ratio of 1.2:1. Tumours of mesenchymal origin are known to cause oncogenic osteomalacia [4]. Hemangiopericytoma is the main described tumour, but other tumour types include fibrous dysplasia, osteosarcoma, chondroblastoma, chondromyxoid fibroma, malignant fibrous histiocytoma and giant-cell tumour. Neurofibromatosis is rarely associated with oncogenic osteomalacia [5]. About 40 cases in the literature report an association between neurofibromatosis and diabetes mellitus. Morlock et al. were the first to measure vitamin D metabolites [6]. Studies have shown that melatonin deficiency may play a role in the pathogenesis of hyperphosphaturia by decreasing sodium–phosphate cotransport, increasing the level of cyclic adenosine monophosphate (cAMP) and the unantagonized effect of dopamine on phosphate reabsorption[7]. Parathyroid overactivity exaggerates the phosphate loss in urine, which is the case in our patient. Given the absence of calcium malabsorption, it could be a primary hyperparathyroidism. Indeed, the association of hyperparathyroidism with parathyroid adenoma and neurofibromatosis is known [8].
Other authors suggest that increased secretion of fibroblast growth factor-23 (FGF-23) by cells from neurofibromas induce phosphorus renal loss. Moreover, augmented FGF-23 plasma levels also inhibit renal 25(OH)-1-α-hydroxylase activity, leading to decreased calcitriol synthesis, thus producing a form of acquired vitamin D resistance. This recent hypothesis suggests that X-linked hypophosphataemic rickets, autosomal dominant hypophosphataemic and tumour-induced osteomalacia (TIO) as well as neurofibromatosis share a common underlying pathophysiological mechanism due to FGF-23 [9].
Metabolic abnormalities in oncogenic osteomalacia are hyphophosphataemia, hyperphosphaturia, low or normal serum calcium, raised alkaline phosphatase, low concentrations of 1,25 dihydroxy vitamin D, decreased tubular resorption of phosphates, normal parathormone levels and low urinary calcium.
In case of neurofibromatosis, clinical examination and laboratory investigations confirm the diagnosis. Dysplastic skeletal lesions are also known to be frequent in neurofibromatosis and are explained by mesodermal dysplasia, are intrinsic to the disease and appear early in life. Even though they cause bone deformities, they are not associated with disturbances in calcium and phosphate metabolism. Radiographs may reveal multiple symmetrical pseudofractures (Looser's zone), loss of trabecular structure, and non-specific decrease in bone radiodensity. Clinical examination and laboratory investigations generally confirm the diagnosis.
In our case, we did not have the facility to measure serum 1,25-(OH) vitamin D, but the reported levels in seven similar cases was normal[5]. Phosphate depletion normally stimulates renal 1-alpha hydroxylation of vitamin D, resulting in marked elevation of serum 1,25-(OH)2 vitamin D concentrations [10]. Thus a normal level of 1,25-(OH) vitamin D in osteomalacia of neurofibromatosis should be regarded as inappropriately low, especially when hypophosphataemia is low.
The treatment is daily administration of phosphorus and 25-OH vitamin D at a pharmacological dose, leading to correction of phosphataemia and the disappearance of bone pain [7].
The clinical resolution of symptoms and normalization of serum biochemical markers of bone turnover such as osteocalcin and alkaline phosphatase activity, tend to take longer. The same result was obtained by Morlock who noted, however, a persistence of hyperphosphaturia[6].
The treatment of choice in oncogenic osteomalacia is tumour removal [3]. If the tumour can be completely removed, then this leads to a dramatic improvement in the clinical course of the disease and biochemical markers, and may be curative. The clinical resolution of symptoms and normalization of serum biochemical markers of bone turnover such as osteocalcin and alkaline phosphatase activity tend to take longer. Therapy is aided by phosphate replacement and the addition of vitamin D and calcium. The scattered nature of neurofibromas in neurofibromatosis makes their surgical excision unrealistic and thus it is impossible to prove a link between tumours and metabolic abnormalities, as has been reported with other removable mesenchymal tumours.
CONCLUSION
Osteomalacia in neurofibromatosis is a very rare entity and generally distinct from the more common dysplastic skeletal effects of this disease. In patients with neurofibromatosis, although numerous neurofibromas are present, it is probably the largest ones or those with recent growth that cause hypophosphataemic osteomalacia, so their surgical removal should be attempted to achieve permanent cure, along with high doses of calcitriol and oral phosphate.