ABSTRACT
Kikuchi-Fujimoto's disease is a self-limiting and rare disorder of unknown aetiology. The typical presentation includes fever, cervical lymphadenopathy and night sweats. Consequently, it is part of the differential diagnosis of infectious, lymphoproliferative and connective tissue diseases. Histology demonstrates necrotizing histiocytic lymphadenitis. Treatment is symptomatic with non-steroidal anti-inflammatory agents, although there are reports of corticosteroid use in complicated cases. We present the case of a 23-year-old woman admitted to hospital for fever and cervical lymphadenopathies, and diagnosed with Kikuchi-Fujimoto's disease.
LEARNING POINTS
- Kikuchi-Fujimoto's disease is an uncommon condition that should be considered in the differential diagnosis of cervical lymphadenopathy.
- Lymph node biopsy is required for the diagnosis and to rule out malignancy.
- Long-term follow-up is important because of an increased risk for recurrence and for the development of systemic lupus erythematosus.
KEYWORDS
Lymphadenopathy, histiocytic necrotizing lymphadenitis, Kikuchi-Fujimoto's disease, glucocorticoid therapy.
INTRODUCTION
Kikuchi-Fujimoto's disease is a benign and self-limited disorder characterized by regional cervical lymphadenopathy with tenderness, usually accompanied by mild fever and night sweats[1,2]. It was described in Japan for the first time in 1972 and in Spain in 1985. The aetiology is unknown, but some authors consider that it can be caused by an immunological reaction secondary to viral infection (Epstein-Barr virus (EBV), cytomegalovirus (CMV), parvovirus B19 or human herpes virus type 6)[3]. We here describe the case of a young woman admitted for fever and cervical adenopathies due to Kikuchi-Fujimoto's disease.
CASE REPORT
A previously healthy 23-year-old woman presented to the emergency department with a 2-month history of enlarged laterocervical adenopathy accompanied by fever of up to 38ºC, fatigue and night sweats. She had started complaining of loss of appetite 1 week before admission to hospital, where a lymphoma was the major concern. She denied loss of weight, maintained her functional baseline, and complained of limitations in the range of motion of her neck due to pain. No chills, myalgias, arthralgias, changes in bowel habits or skin rashes were mentioned. Furthermore, she denied vision impairment, hearing deficits or dizziness. She did not report dyspnoea, cough, or chest or abdominal pain, and there was no abdominal fullness. She had not recently travelled outside Spain and had not been exposed to animals. The patient had no significant past medical story and was not taking any medication. She did not smoke cigarettes or drink alcohol. She was an administrative student with a family history of leukaemia and pancreatic cancer.
Physical examination revealed a healthy appearing young woman, with a significant 4 cm left laterocervical node and 3 cm supraclavicular bilateral adenopathies tender to palpation, non-erythematous and without surrounding cellulitis. No axillary, epitrochlear, inguinal or popliteal lymphadenopathy was detected. She was afebrile and showed no other significant physical findings.
Laboratory data revealed an erythrocyte sedimentation rate of 87 mm/h (reference range, 0–20) and a C-reactive protein of 25.94 mg/l. White cell count was 2,600/mm3 (neutrophils 47.4%, lymphocytes 41.3%, monocytes 11%), platelets were normal, and AST and ALT were 45 and 49 IU/l, respectively. Lactic acid dehydrogenase was 463 U/l and antinuclear antibody screen was positive at 1:160 (with negative anti-DNA). The peripheral blood smear showed leukopenia without atypical cells. Blood cultures were negative. The tuberculin skin test (PPD) was non-reactive after 72 h. The beta-2-microglobulin level was 2.1 μg/ml. EBV, CMV, herpes virus type 6 and parvovirus B19 antibody tests were negative. Chest X-ray and thoraco-abdominal CT disclosed neither systemic lymphadenopathy nor other abnormalities. A neck ultrasound and magnetic resonance showed pathological left laterocervical lymph nodes 2 cm in diameter. Further tests, including echocardiography and urine culture, performed to rule out other causes of fever, were negative. The patient underwent an excisional lymph node biopsy. Pathological examination (Figs. 1 and 2) revealed a necrotizing paracortical lymphadenitis with CD68+ macrophages, plasmacytoid CD123+ cells, and CD3+ and CD8+ lymphocyte predominance, consistent with Kikuchi-Fujimoto's disease.
The patient was treated with non-steroidal anti-inflammatory drugs (NSAIDs) and corticosteroids followed by disease resolution.
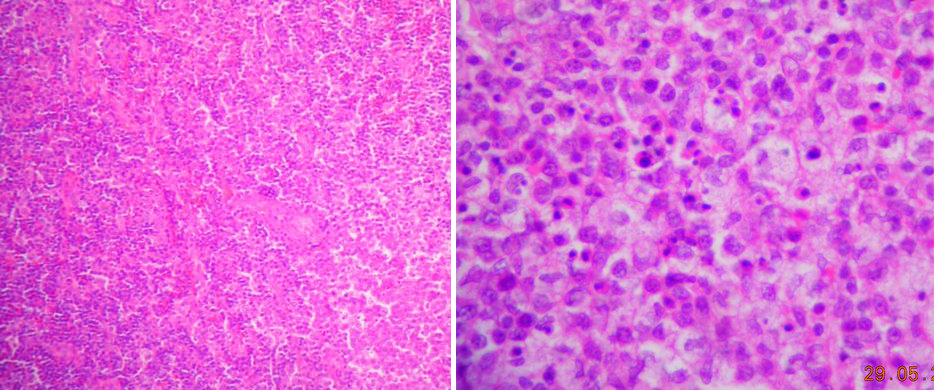
Figure 1 (click to enlarge)
Figure 1: Hematoxylin eosin stains (40x and 400x) show a developed focus of necrosis with numerous apoptotic bodies close to macrophages and histiocytes.
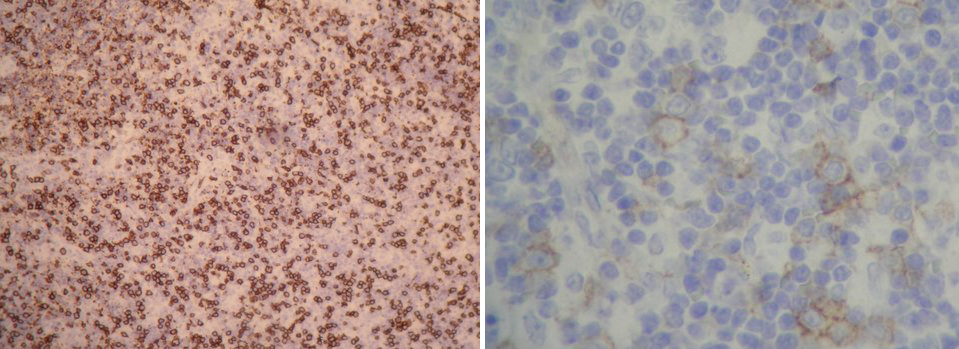
Figure 2 (click to enlarge)
Figure 2: Immunohistochemical staining (40x and 400x) show that the focus of necrosis is in a T lymphocites area (CD3 and CD8) with abundant CD68 macrophages, some CD123 cells and scarce B lymphocites (CD20).
DISCUSSION
The broad differential diagnosis of cervical lymphadenopathy in an adult includes cancer (head and neck cancer, metastatic cancer, Hodgkin's and non-Hodgkin's lymphoma, and Castleman's disease), infections (EBV infection, CMV infection, rubella, tuberculous and non-tuberculous mycobacterial lymphadenitis, toxoplasmosis, cat-scratch disease and AIDS-related lymphadenopathy), autoimmune diseases (systemic lupus erythematosus (SLE) and other conditions (sarcoidosis, Kimura's disease and Kikuchi-Fujimoto's disease)[1]. In a European context, cancer and benign tumours are the most likely diagnoses, followed by infections, SLE and sarcoidosis[4]. The diagnostic work-up usually involves:
- history and clinical examination, including age and time course, signs or symptoms suggesting infection or malignancy, exposures, medications, presence of splenomegaly, localized or generalized lymphadenopathy, and size, consistency, fixation and tenderness of the lymph nodes;
- complete blood count that may point towards leukaemias and lymphomas, or provide useful data for the diagnosis of EBV, CMV, pyogenic infections or immune cytopenias such as SLE;
- serological studies if the history is suggestive of EBV, CMV, HIV, toxoplasmosis, brucellosis or SLE;
- chest X-ray to detect pulmonary infiltrates and mediastinal lymph nodes as well as potential involvement of the lung parenchyma;
- other imaging techniques (CT scan, MRI and ultrasound) employed to differentiate between malignant and benign lymphadenopathy;
- a biopsy whenever there is suspicion of malignancy. If history and findings suggest a benign aetiology, monitoring for 2–4 weeks is advisable. Fine needle aspiration should not be the first intervention and is reserved for the confirmation of cancer relapse in patients with a known primary tumour.
In this case, where a young Spanish woman under 30 years of age with no apparent exposure to infections, presents with symptoms and a family history of malignancy, the first step is to rule out lymphoma. Nevertheless, a physical examination showing localized cervical tenderness with soft lymphadenopathy mitigated against such diagnosis. A normal peripheral smear and a chest X-ray, thoraco-abdominal CT and neck MRI without any other findings made a malignant disease unlikely. Localized infections should have become apparent by history and physical examination, while serological studies, blood and urine cultures, and the PPD test were negative, making an infectious cause unlikely. Therefore the most probable diagnostic option was an autoimmune disease. Patients with SLE can present with a variety of symptoms and signs, but despite a positive ANA titre, this woman did not meet the criteria for SLE. Sarcoidosis often involves hilar lymphadenopathy and lung tissue, presenting with respiratory symptoms. Kimura's disease is a benign, rare, chronic inflammatory disorder which usually presents with painless lymphadenopathy in the head and neck. The clinical features of Kikuchi-Fujimoto's disease are consistent with this patient´s presentation, although it is a very uncommon condition. The results of lymph node biopsy were diagnostic for Kikuchi-Fujimoto's disease.
Kikuchi-Fujimoto's disease is a rare, benign and self-limiting disorder. The aetiology is unknown, but some authors contend that it can be caused by an immunological reaction secondary to viral infection[3]. It is more frequent in young women, presenting with painful laterocervical and supraclavicular adenopathy[2]. In addition, 30–50% of patients with Kikuchi-Fujimoto's disease may have fever, usually low-grade, associated with upper respiratory symptoms[3]. Less frequent symptoms include weight loss, nausea, vomiting, sore throat, night sweats, arthralgias, myalgias and skin rashes[2]. Leukopenia can be found in up to 50% of cases. Atypical lymphocytes in the peripheral blood have also been observed. Light thrombocytopenia, neutropenia and hypertransaminasaemia with elevated lactic acid dehydrogenase have also been reported.
The diagnosis of Kikuchi-Fujimoto's disease requires an excisional biopsy of affected lymph nodes, with findings showing a reactive hyperplasia with circumscribed paracortical foci of necrosis and abundant cellular detritus, surrounded by CD4 lymphocytic and CD68 histiocytic aggregates, with few neutrophils and plasma cells[5,6]. According to Kuo[7], the histopathological features can be classified into three stages: proliferative, necrotizing and xanthomatous. Sometimes, because of similar clinical and histological features, it is problematic to differentiate Kikuchi-Fujimoto's disease from lymphadenitis associated with SLE[8], so the latter should be ruled out.
Treatment of Kikuchi-Fujimoto's disease is symptomatic, requiring the use of analgesics, antipyretics, NSAIDs and, rarely, corticosteroids. Indications for corticosteroid use include the following[6,9]: neurological involvement (aseptic meningitis, cerebellar ataxia), hepatic involvement with elevated LDH levels, and severe lupus-like syndrome with positive ANA titres. Corticosteroid therapy has also been recommended by some authors to shorten the duration of fever[9,10]. Spontaneous recovery occurs in 1–4 months3. Approximately 3–4% of patients have recurrence of the disease, sometimes even several years after the initial episode[3,6]. Patients with Kikuchi-Fujimoto's disease should be followed up for several years to check for subsequent development of SLE[2].
In our case, after a follow-up period of 2 years the patient remained asymptomatic without SLE manifestations.
CONCLUSION
Kikuchi-Fujimoto's disease, although rare, should be part of the differential diagnosis for tender cervical lymphadenopathy. Recognition of this disorder is crucial because it can be mistaken for malignant lymphoma, leading to extensive investigations and the institution of toxic and aggressive treatment. Excisional biopsy of the affected lymph nodes is the gold standard for making an accurate diagnosis. Although Kikuchi-Fujimoto's disease is self-limiting, and only requires symptomatic treatment, long-term follow-up is important to rule out recurrences or the development of autoimmune diseases such as SLE.