2 Internal Medicine Department, Universidad de Antioquia, Medellìn, Colombia
3 Clinical Oncology, National Cancer Institute, Bogotà, Colombia
Accepted: 05/07/2021
Published: 05/08/2021
How to cite this article: Forero Saldarriaga S, Vallejo C, Urrea Pineda L, Osma A, Bonilla Gonzalez C. Gorham-Stout disease with clinical response to sirolimus treatment. EJCRIM 2021;8 doi:10.12890/2021_002740.
Conflicts of Interests: The authors declare there are no competing interests.
This article is licensed under a Commons Attribution Non-Commercial 4.0 License
ABSTRACT
Gorham-Stout disease (GSD) is a rare clinical entity of unknown aetiology, with osseous resorption, vascular structure proliferation and inflammation of adjacent soft tissues. The clinical spectrum varies from asymptomatic patients to patients with pain, functional limitation or deformity. GSD may affect 1 or multiple bones in any location; however, predilection for the maxillofacial area and upper limbs has been described. We present the case report of a 33-year-old male patient with facial pain and loss of dental pieces; imaging showed extensive lytic involvement of the mandible and cranial bones. After exclusion of malignant, metabolic, infectious, or autoimmune disease, a diagnosis of GSD was made. Treatment with sirolimus was prescribed, achieving a symptomatic improvement and stability of imaging findings. The importance of the knowledge of this orphan disease is highlighted, to achieve a prompt diagnosis and medical treatment.
LEARNING POINTS
- Gorham-Stout syndrome constitutes an orphan disease with an unknown aetiology, it must be considered in young patients with osteolysis of unknown cause. .
- The diagnosis of Gorham-Stout syndrome is an exclusion one.
- Drugs with antiproliferative and antiangiogenic activity such as mTOR inhibitors are an attractive option for the management of this disease.
KEYWORDS
Gorham disease, osteolysis, sirolimus, lymphangiomatosis
CASE DESCRIPTION
AA 33-year-old male with a personal history of cigarette smoking of 5 packs/year, with no family history of cancer. In November of 2015, the patient debuted with intense pain in the left maxillary region; initially, a periapical dental abscess was suspected, antibiotic therapy was prescribed and the dental piece was extracted with no improvement. Thereafter, purulent material was observed in the base of a dental piece, and various dental pieces were lost spontaneously, which led to the diagnosis of oroantral fistula. Over 4 years, until 2019, the patient underwent multiple surgical interventions in an effort to close the oroantral fistula, with partial symptomatic improvement of short duration. In February 2019, computed tomography (CT) of the paranasal sinuses was performed, showing osteolysis of the posterolateral wall of the left maxillary sinus and homolateral papyraceous lamina (Fig. 1), osteolysis of the lateral wall of the left orbit (Fig. 2), and osteolysis of the squamous portion of the left temporal bone (Fig. 3). CT 3D reconstruction depicted osteolysis of the left superior maxillary and mandibular bone (Fig. 4).
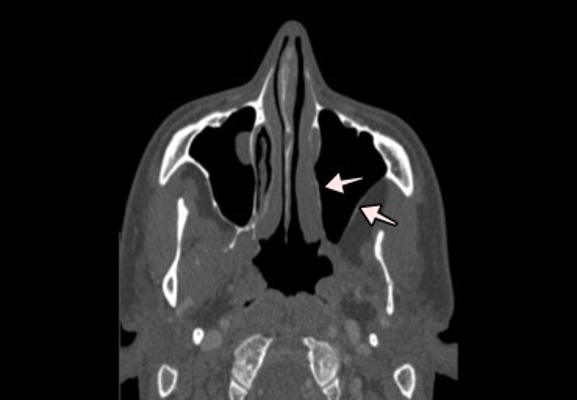
Figure 1 (click to enlarge)
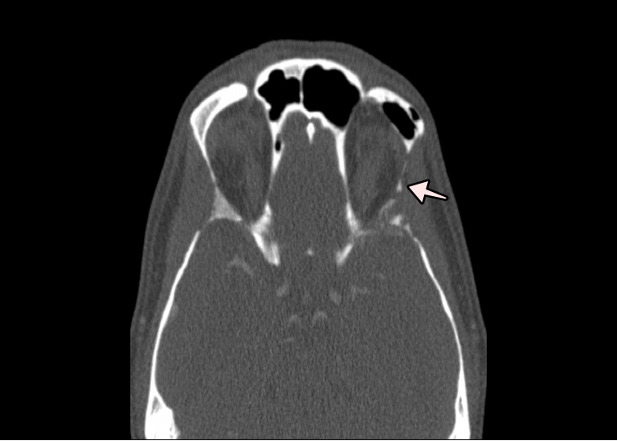
Figure 2 (click to enlarge)
Figure 1. Face and paranasal sinus CT. Osteolysis of the posterolateral wall of the left maxillary sinus and homolateral papyraceous lamina. The arrows highlight the osteolytic defects
Figure 2. Face and paranasal sinus CT. Osteolysis of the lateral wall of the left orbit. The arrow highlights the osteolytic defects
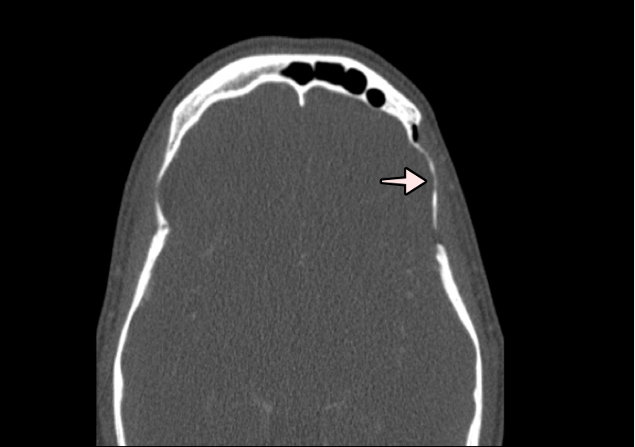
Figure 3 (click to enlarge)
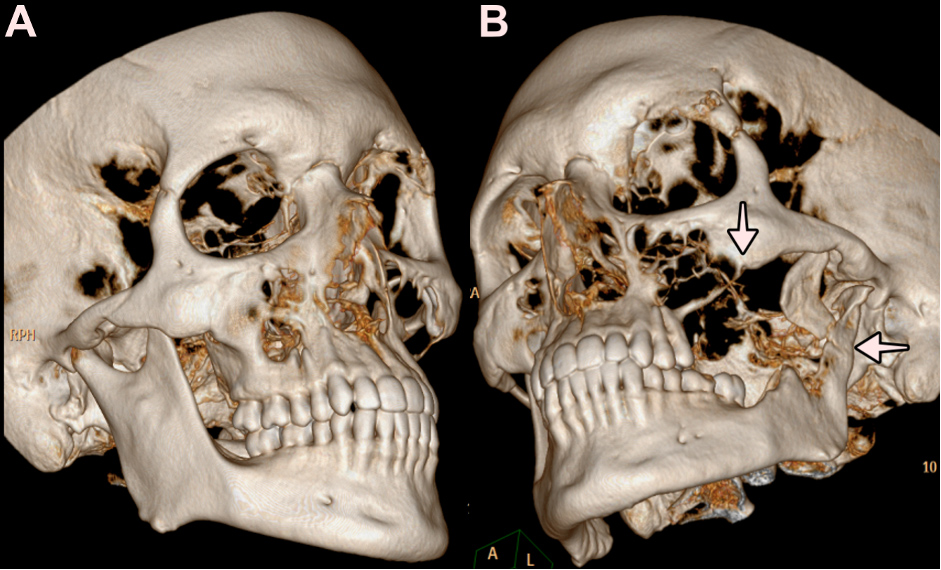
Figure 4 (click to enlarge)
Figure 3. Face and paranasal sinus CT. Osteolysis of the squamous portion of the left temporal bone. The arrow highlights the osteolytic defects
Figure 4. Face and paranasal sinus CT with 3D reconstruction. A. 3D reconstruction with a right superior maxillary and mandibular bone of normal aspect. B. 3D reconstruction with osteolysis of the left superior maxillary and mandibular bone. The arrows highlight the osteolytic defects
Multiple studies were performed, including thorax, abdomen and pelvis CT, which yielded no pathological findings. In March 2019, a maxillary bone biopsy was performed which showed non-specific dilated vascular and lymphatic channels with no malignancy criteria. In the differential diagnosis, multiple myeloma, malignant histiocytosis, metastatic malignant disease, metabolic pathology, infectious or autoimmune diseases were considered; however, all these possibilities were ruled out after the thorough analysis of imaging, laboratory tests and the histopathology. After correlation of these diagnostic procedure results with clinical features, the diagnosis of Gorham-Stout disease (GSD) was established. The patient underwent treatment with a single 4 mg dose of zoledronic acid but symptomatic worsening was observed, with maxillary pain progression. Subsequently, the patient attended the Instituto Nacional de Cancerología for a second opinion; a multidisciplinary medical board was performed and the patient was considered a poor candidate for surgical or radiotherapy treatment given the extensive mandibular, orbital and cranial involvement. Hence, medical therapy with sirolimus 1 mg BID was started. Such treatment was given from November 2019 up to the date of this manuscript, with good clinical response, no associated toxicity, symptom improvement and stable imaging findings.
DISCUSSION
GSD, also known as ghost bone disease or vanishing bone disease, is a rare clinical entity of benign histopathological behaviour, known for the proliferation of vascular structures which result in massive bone osteolysis. Clinical presentation is variable, characterized by pain, loss of function in limbs and in rare occasions some catastrophic features such as massive chylothorax possibly related to thoracic lymphatic dysfunction. Approximately 30% of cases affect the maxillary region as in our patient; however, any bone may be eventually affected – other than the maxillary region, predilection for upper limb bones and the pelvis has been described [1, 2]. Even though this clinical condition may be observed at any age, it generally presents in the second or third decade, with no gender, race or geographical predilection. Clinical courses are heterogeneous, from patients with spontaneous remission to those with an aggressive disease which rapidly progresses to death [2].
To date, the pathophysiology is not clear. Some authors highlight that bone resorption occurs due to increased osteoclast activity mediated by T lymphocytes, interferon-gamma, tumour necrosis factor, prostaglandin E2, interleukin-1 and interleukin-6 [1, 2]. This inflammatory response induces the receptor activator of nuclear factor-kappa B ligand (RANKL), binding to the RANK receptor expressed in osteoclast precursors which finally leads to differentiation and osteoclast activation and osteoblast inhibition [2]. Macrophages develop an important role, as they may induce osteoclast activity and stimulate lymphangiogenesis through the production of VEGF types A, C and D. These growth factors are important in the lymphatic vessel invasion and derive in the mTOR (mechanistic target of rapamycin) pathway activation, which is crucial in cellular proliferation and growth [2].
GSD diagnosis is challenging due to its extremely rare incidence, and non-specific clinical features. Therefore, plenty of differential diagnoses such as infectious diseases, cancer, inflammation and endocrine diseases must be ruled out first [1, 2]. Heffez et al. described some diagnostic criteria to consider GSD [3], all of which were present in our patient (Table 1).
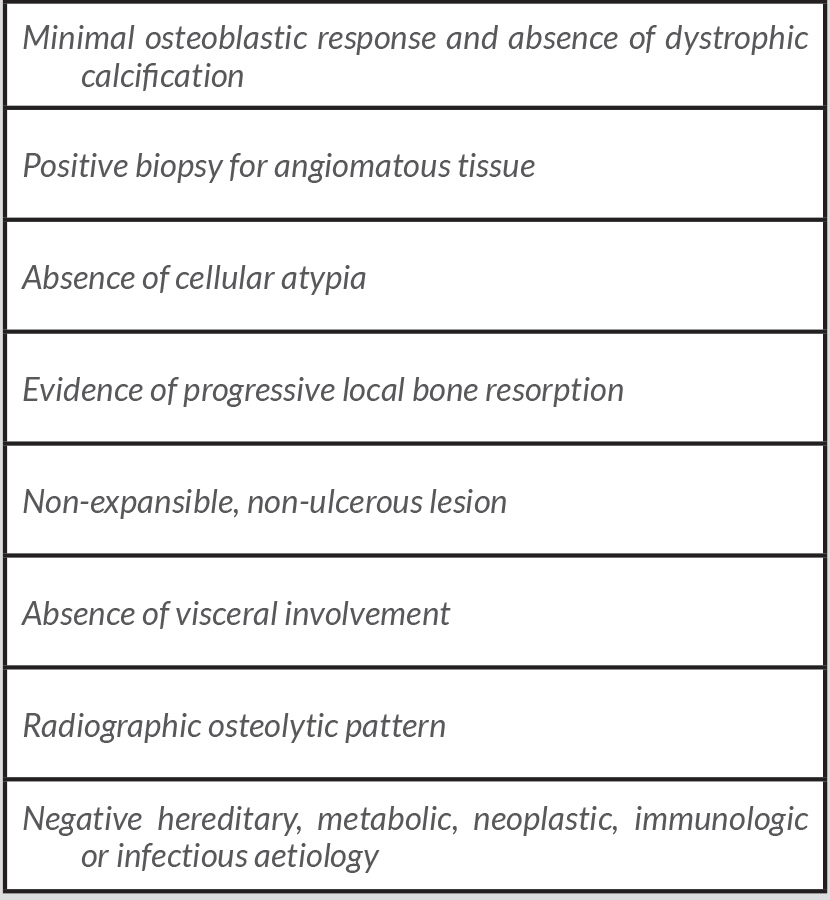
Table 1 (click to enlarge)
Table 1. Diagnostic criteria for massive osteolysis (adapted from Heffez et al.,1983)
Due to the lack of knowledge of the disease, no standard treatment has been described. Conventional therapy includes the local inhibition of bone resorption with drugs such as bisphosphonates, denosumab and calcitonin; however, the real efficacy of these treatment regimens has not been backed by large-scale studies. mTOR inhibitors constitute an attractive treatment option given their inhibitory activity on cellular proliferation and angiogenesis, with an acceptable safety profile [4]. In fact, case reports with satisfactory use of these drugs are becoming increasingly frequent [4]. In our case, sirolimus 1 mg PO BID was prescribed, according to previous case reports available; with this dose, our patient presented clinical benefit with no imaging progression. The local management with surgery or radiotherapy is reserved for refractory disease or rapidly progressive disease, including those patients with pathologic fractures [5].
- Nikolaou VS, Chytas D, Korres D, Efstathopoulos N. Vanishing bone disease (Gorham-Stout syndrome): a review of a rare entity. World J Orthop 2014;5(5):694–698.
- SKeyser CE, Saltzherr MS, Bos EM, Zillikens MC. A large skull defect due to Gorham-Stout disease: case report and literature review on pathogenesis, diagnosis, and treatment. Front Endocrinol (Lausanne) 2020;11:37.
- Heffez L, Doku HC, Carter BL, Feeney JE. Perspectives on massive osteolysis. Report of a case and review of the literature. Oral Surg Oral Med Oral Pathol 1983;55(4):331–343.
- Freixo C, Ferreira V, Martins J, Almeida R, Caldeira D, Rosa M, et al. Efficacy and safety of sirolimus in the treatment of vascular anomalies: a systematic review. J Vasc Surg 2020;71(1):318–327.
- Fares MY, Fares J, Fares Y, Abboud JA. Gorham-Stout disease of the shoulder: clinical, pathologic and therapeutic considerations. Arch Bone Jt Surg 2020;8(1):58–69.