ABSTRACT
Type 1 Gaucher disease (GD) is a rare autosomal recessive lysosomal storage disorder caused by deficient activity of beta-glucocerebrosidase, leading to accumulation of its substrate (glucosylceramide) in macrophages of the reticuloendothelial system, which are then referred to as Gaucher cells. The most frequent symptoms are asthenia, spleen and liver enlargement, bone abnormalities and cytopenia due to bone marrow infiltration. Lung involvement in GD is a rare finding, and it is unclear whether it may regress under enzyme replacement therapy (ERT) or substrate reduction therapy (SRT). Here we report a case of type 1 GD recently diagnosed in an elderly patient complicated by infiltrative lung disease, which responded to ERT.
LEARNING POINTS
- Type 1 Gaucher disease (GD) is the most common type of GD diagnosed in adulthood, and is characterized by great clinical heterogeneity.
- Pulmonary involvement is rare, mostly characterized by infiltrative lung disease and pulmonary hypertension, and typically unresponsive to enzyme replacement therapy (ERT).
- Type 1 GD should be included in the differential diagnosis of infiltrative lung disease of unclear origin in patients with cytopenia and/or splenomegaly.
- Infiltrative lung disease due to type 1 GD may respond to ERT even in elderly patients.
KEYWORDS
Gaucher disease, infiltrative lung disease, enzyme replacement therapy
CASE DESCRIPTION
A 79-year-old man of Portuguese ancestry with high blood pressure treated with enalapril was hospitalized for partial epileptic seizures. Brain imaging was in favour of cerebrovascular leukopathy. Complete blood count and serum chemistry revealed anaemia, thrombocytopenia and hyperferritinaemia. Bone marrow aspiration showed infiltration with lipid-laden histiocytes. Abdominal ultrasound and a CT scan disclosed an enlarged liver and spleen (16 cm), and pulmonary infiltrates. The patient was referred to our tertiary centre for suspected GD.
Physical examination found hepato-splenomegaly and akinetic-hypertonic Parkinsonism without resting tremor. The patient reported mMRC grade 2 dyspnoea. Cardiac and pulmonary examination was normal, without any auscultatory crackles or signs of heart failure. Laboratory tests confirmed thrombocytopenia (95 G/l), anaemia (11.5 g/dl with a MCV of 98 fl), lymphocytopenia (0.57 G/l) and hyperferritinaemia (2150 µg/l). C-reactive protein was slightly elevated (9.2 mg/l), the CDK-EPI estimated glomerular filtration rate was altered (48 ml/min/1.73m2) and the serum protein electrophoretic profile was normal. Chitotriosidase was markedly elevated (4735 nmol/h/ml, N <120), as was lyso-hexacylceramide (74.8 nmol/l, N <2.0). The diagnosis of GD was confirmed by reduced beta-glucocerebrosidase activity (0.93 µmol/l/h, N 3.36–13.2) and genetic analysis which showed a homozygous mutation (p.N370S) in the glucocerebrosidase (GBA) gene.
Osteodensitometry and bone x-rays showed osteopenia but no typical Gaucher bone involvement or osteoporotic fractures. The infiltrative lung disease (ILD) remained unexplained, although compatible with GD involvement, while autoimmune diseases, allergic alveolitis and infection were excluded by extensive biological work-up. Pulmonary function tests were inconclusive due to the patient’s lack of respiratory coordination. Fibre-optic bronchoscopy and bronchoalveolar lavage were considered too invasive in view of the patient’s age and were not performed. A control chest CT scan was performed 3 months later, and showed stable ILD (Fig. 1). Spirometry was again not contributive.
The decision to start enzyme replacement therapy (ERT) despite rather mild disease in an elderly patient was taken with the advice of the French Referral Center for Lysosomal Diseases, due to possible pulmonary involvement and slight bone infiltration of the femoral and humoral heads on whole body MRI. The patient agreed to receive imiglucerase (CEREZYME®, 3600 IU every 2 weeks), tolerance to which proved excellent. Biological parameters improved markedly: after 8 months of treatment, a 2-fold decrease in ferritinaemia and a 20-fold decrease in chitotriosidase (267 nmol/h/ml) were observed. Splenomegaly had regressed (13 cm). However, Parkinson disease required treatment with levodopa/benserazide 50/12.5 mg three times a day. At 18-month follow-up, the patient reported a better general status, less dyspnoea and increased exercise tolerance, while the blood count had normalized (haemoglobin 13.6 g/dl, platelets 181 G/l) except for lymphopenia (0.7 G/l).
Chest CT scan was checked at 18-month follow-up and revealed regression of infiltrative lung lesions (Fig. 1). Unfortunately, the functional improvement experienced by the patient could not be quantified because of his inability to perform spirometry. Given this favourable outcome under ERT, the ILD was finally attributed to type 1 GD.
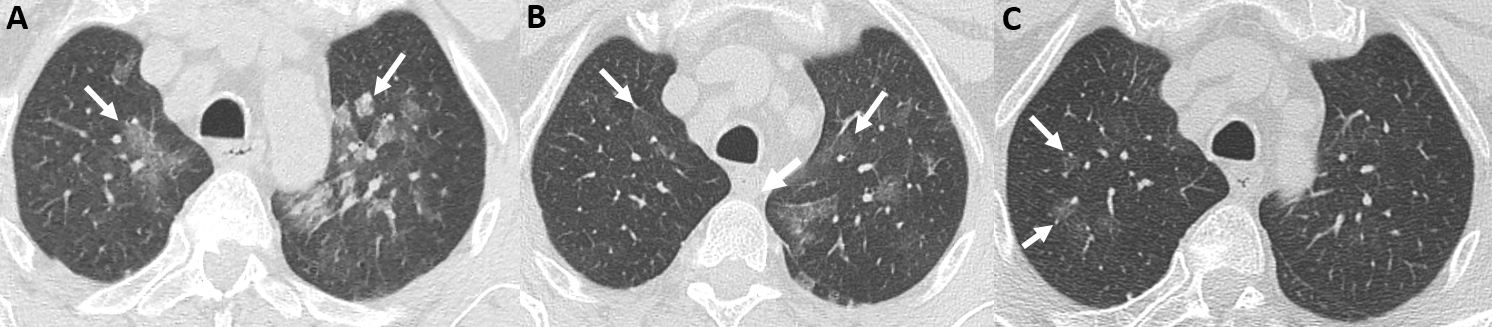
Figure 1 (click to enlarge)
Figure 1. Chest CT images in the axial plane centred on the upper lobes. (A) On 10 September 2019, imaging shows multifocal large ground-glass opacities (white arrows) without condensation or micronodules. (B) On 14 February 2020, diffuse ground-glass opacities were resolved partially in both lungs (white arrows). (C) On 21 December 2020, imaging shows almost complete disappearance of the parenchymal abnormalities, with persistent small ground-glass opacities in the right upper lobe (white arrows)
DISCUSSION
Gaucher disease is the most frequent lysosomal disease. It is due to an autosomal recessive mutation of the gene coding for beta-glucocerebrosidase, leading to accumulation of glucosylceramide (a cellular membrane component) in macrophages, which are then referred to as Gaucher cells. Type 1 GD, without neurological damage, is responsible for around 90% of adult cases. Type 2 and type 3 include severe neurological damage occurring early in life. The clinical spectrum of type 1 GD is characterized by extreme clinical heterogeneity, ranging from asymptomatic to more severe presentations. The median age at the time of diagnosis is 22 years (0–84) in the French cohort [1]. The most frequent symptoms and signs leading to the diagnosis are asthenia, splenomegaly and thrombocytopenia [1]. Frequent complications involve the bones (infarction, aseptic osteonecrosis or fractures), spleen (splenic infarction or splenic rupture leading to splenectomy), central nervous system (Parkinsonism) and immune system (monoclonal gammopathy of undetermined significance) [1]. Fatigue is fairly common and debilitating. Diagnosis is based on demonstration of a loss of beta-glucocerebrosidase activity (by serum assay or on blotting paper). Other arguments are demonstration of Gaucher cells on bone marrow aspiration or any other tissue, and ultimately GBA gene mutation analysis. ERTs (imiglucerase, velaglucerase and taliglucerase) are the reference treatment for type 1 GD. Although extremely expensive, ERTs have been approved by the European Medicines Agency and are granted orphan drug status in many countries. Published cohort studies and registries have shown that ERTs give effective control of the non-neurological symptoms and signs of GD, with an increase in the number of red blood cells and platelets, a decrease in the size of the liver and spleen, and a decreased likelihood of bone events. Their most common side effects are hypersensitivity reactions, associated with dyspnoea, coughing, urticaria or angioedema in less than 1% of patients. In France, the decision to offer ERT or substrate reduction therapy (SRT) to patients with GD must be based on advice from a multidisciplinary panel of experts from the French Referral Center for Lysosomal Diseases, and approximately 60% of GD patients are being treated long term. Criteria for treatment include symptomatic thrombocytopenia or anaemia, and radiological bone involvement [2]. Unfortunately, most complications will not regress even though adapted treatment is started.
Although this 79-year-old patient presented with epilepsy and cerebral leukopathy, these neurological findings were attributed to chronic hypertension: we excluded type 3 GD and considered him as a case of type 1 GD complicated by early Parkinson disease. A pauci-symptomatic ILD without any obvious aetiology was documented, and although pulmonary involvement is an exceptional complication of type 1 GD, we believe that the unexpected favourable outcome of ILD under ERT could be viewed as an argument for GD being a plausible cause.
While common in some other lysosomal storage diseases such as Niemann-Pick disease [3], pulmonary complications are rare in type 1 GD. The largest studies to date of pulmonary involvement in GD found a prevalence of 1.9% [4] and 3.33% (5/150) [5], while no case was reported in the French GD registry [1]. Pulmonary involvement in GD is mostly characterized by ILD and pulmonary hypertension, and is typically associated with other severe damage. The imaging pattern of ILD due to GD includes alveolar and interstitial opacities with a predominantly lymphatic distribution, sometimes combined in a mosaic pattern, and enlarged hilar and mediastinal lymph nodes [4–6]. Improvement in pulmonary damage with ERT has been reported in a few cases but is uneven and consistently below the expected result for other types of impairment. There may be regression of dyspnoea or improvement in pulmonary function tests, but there is typically no normalization of parenchymal involvement on CT scan [4-5]. A role for ERT in preventing such damage has not been demonstrated, and the possibility that it might trigger or aggravate pulmonary hypertension has been raised [4-5].
In our case, ILD was incidentally discovered in a pauci-symptomatic 79-year-old patient, without severe GD damage except for Parkinson disease, and responded to ERT. Pulmonary involvement might remain underdiagnosed or under-reported in elderly untreated GD patients. We suggest that type 1 GD should be included in the differential diagnosis of ILD of unclear origin in patients of any age with cytopenia and/or splenomegaly.