ABSTRACT
Background: Patients with sickle cell disease can experience various crises including sequestration crisis, haemolytic crisis and aplastic crisis. Due to alloantibody formation, transfusion alloantibodies can cause a haemolytic crisis. Treatment involves avoiding packed red blood cell transfusions, as well as intravenous immunoglobulin, steroids and eculizumab to decrease the chances of haemolysis.
Case description: We report the case of a 42-year-old man who was found to have worsening anaemia after packed red blood cell transfusion with evidence suggestive of haemolytic crisis. Due to reticulocytopenia, aplastic crisis was also suspected and later confirmed via parvovirus IgG and IgM titres. The patient did not improve with steroid and intravenous immunoglobulin therapy and was treated with eculizumab as a salvage therapy.
Conclusion: Concurrent hyper-haemolytic crisis and aplastic crisis should be suspected in patients with features of haemolysis and reticulocytopenia. Prompt recognition and treatment with eculizumab are paramount in those who fail steroid and intravenous immunoglobulin treatment.
LEARNING POINTS
- Treatment of hyper-haemolytic and aplastic crisis in sickle cell disease with eculizumab offers therapeutic benefit.
- A high index of suspicion for hyper-haemolytic crisis and aplastic crisis should be maintained in those with haemolytic features as well as reticulocytopenia in the setting of sickle cell disease.
KEYWORDS
Sickle cell disease, hyper-hemolysis, eculizumab, aplastic anemia
INTRODUCTION
Sickle cell disease is a chronic multisystem disease characterized by vaso-occlusive phenomena and haemolysis [1]. These clinical manifestations are due to a point mutation in the beta globin gene that causes haemoglobin polymerization [1, 2]. The most common cause for hospital admissions is painful vaso-occlusive phenomena [3]. Other clinical complications include acute chest syndrome, pulmonary hypertension, avascular necrosis, heart disease and renal failure [4, 5].
Sickle cell disease is known to produce a chronic haemolytic anaemia that can acutely worsen with blood transfusion [5, 6]. The major precipitating factors for acute anaemia are aplastic crisis, splenic sequestration crisis and hyper-haemolytic crisis [6, 7]. Alloimmunization may occur as a result of transfusions in patients being treated for acute haemolytic crisis [6]. Delayed hypersensitivity transfusion reactions and hyper-haemolytic syndrome must be readily recognized as further packed red blood cell transfusions can increase mortality in these patients [6, 7]. We present the case of a 42-year-old man with sickle cell disease who developed acute anaemia and reticulocytopenia following packed red blood cell transfusion which improved following eculizumab therapy.
CASE DESCRIPTION
A 42-year-old man with a medical history of sickle cell disease, complicated by avascular necrosis of bilateral hips, and rheumatoid arthritis presented to the hospital with complaints of fevers, fatigue and dyspnoea for 1 week. He reported no chest pain, joint pain, cough or sick contacts. On admission he was febrile to 38.3°C, had a heart rate of 102 beats per minute, and pulse oximetry of 95% on room air. Physical examination noted conjunctival pallor and mild rales in bilateral lung fields. Chest x-ray showed mild bilateral interstitial infiltrates. Laboratory analysis revealed a haemoglobin of 5.3 g/dl (normal range 13.2–17.5 g/dl) with an absolute reticulocyte count of 0.043 µl (normal range 0.010–0.110 µl) and a white blood cell count of 8.0 (normal range 4.5–11 k/µl). The patient was started on vancomycin and piperacillin-tazobactam after blood cultures were obtained and an intravenous fluid bolus was given for possible sepsis. Parvovirus IgG and IgM titres were also obtained due to concern of aplastic crisis. He was transfused with two units of packed red blood cells and was started on erythropoietin due to reticulocytopenia. Post-transfusion haemoglobin was 5.0 g/dl. Typing and cross-matching revealed antibodies to blood products.
Due to an inadequate response to transfusions, haemolytic work-up was pursued (Table 1).
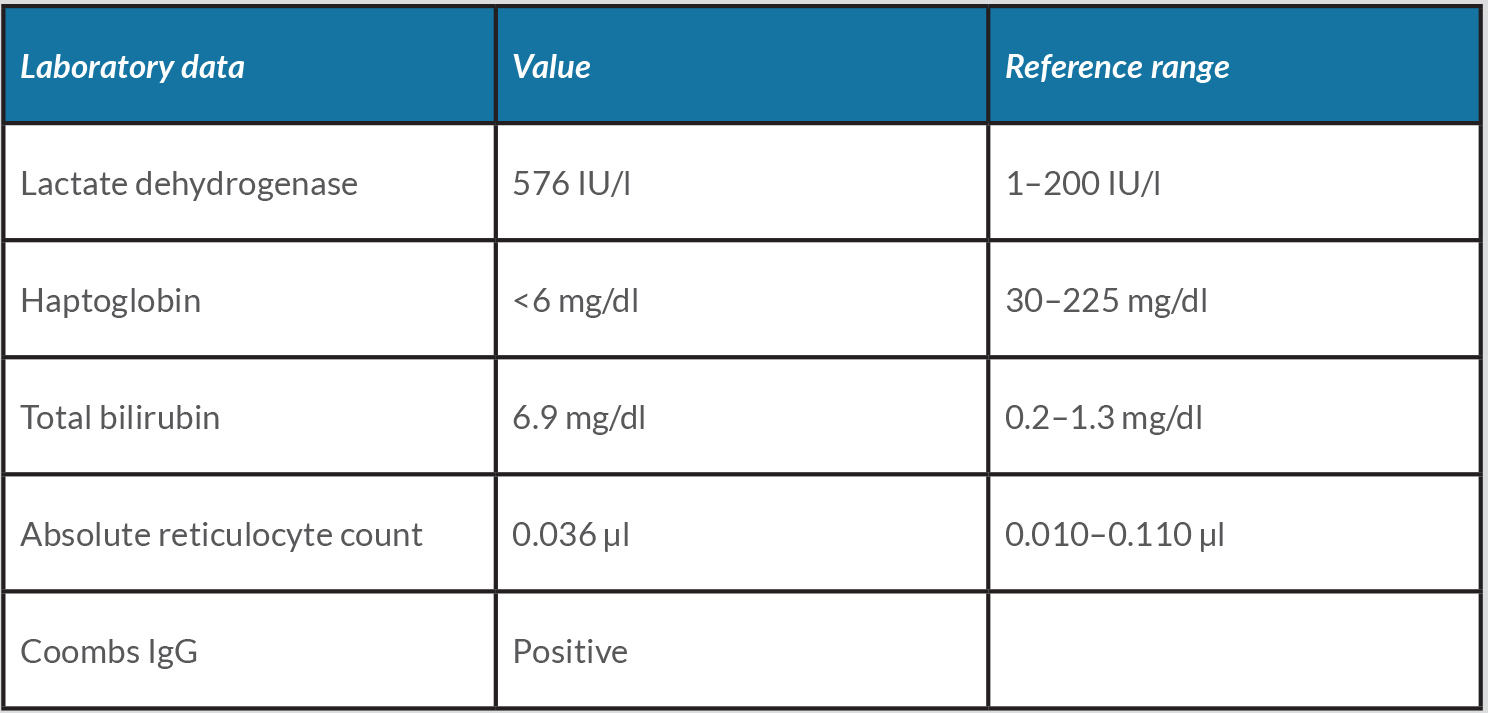
Figure 1 (click to enlarge)
Table 1. Haemolytic work-up values
The patient was started on methylprednisolone and intravenous immunoglobulin for 5 days. He subsequently developed worsening fatigue, jaundice and hypoxia. Haemoglobin remained low despite corticosteroid and intravenous immunoglobulin therapy. Eculizumab 900 mg was then started. Subsequent parvovirus IgG and IgM titres were elevated, suggesting concurrent aplastic crisis.
After initiation of eculizumab, the patient’s clinical status improved. He was subsequently discharged on a prednisone taper and haematology referral.
DISCUSSION
Acute transfusion reactions usually occur during or immediately after transfusion [8]. Symptoms may include chills, fever, hypotension, haemoglobinuria and renal failure. Complications like delayed hypersensitivity transfusion reactions with hyper-haemolysis occur 5–20 days after transfusion [9].
Hyper-haemolysis is a rare but often a fatal complication of sickle cell disease. It is characterized by severe anaemia with a post-transfusion decrease in haemoglobin [10]. Findings include destruction of donor and recipient packed red blood cells, low reticulocyte counts, and negative Coombs antibody assays [8]. Our patient developed aplastic anaemia from acute parvovirus infection, with hyper-haemolytic crisis from a delayed transfusion reaction.
Multiple mechanisms have been postulated as the cause of hyper-haemolysis including bystander red blood cell haemolysis via antibody formation against human leukocyte antigen protein and complement pathway, and macrophage-mediated destruction of red blood cells [8]. Bystander haemolysis is thought to be due to formation of antigens of non-red blood cells leading to complex formation with red blood cells, making sickle cells more vulnerable to complement-mediated destruction [8]. Macrophage-mediated hyper-haemolysis is thought to be due to macrophage interaction with vascular cell adhesion molecule-1 molecules on sickle reticulocytes [8].
The management of hyper-haemolysis includes conservative transfusion strategies as additional transfusions may lead to worsening haemolysis [8]. Our patient was managed conservatively after transfusion with three units of packed red blood cell due to concern for additional haemolysis as well as due to lack of compatible packed red blood cell availability. Intravenous immunoglobulin therapy has been proposed to reduce haemolysis by accelerated clearance of endogenous pathogenic auto-antibodies and inhibition of components of the complement cascade [12]. Corticosteroids are also used to reduce autoimmune-mediated haemolysis [8]. Rituximab has also been used for the treatment of delayed transfusion reactions [12, 13].
Eculizumab has been used successfully to treat delayed hypersensitivity transfusion reaction. As a C5-convertase inhibitor, it inhibits the complement cascade. Treatment with two doses of eculizumab was used in three patients reported by Dumas et al. [14]. Boonyasampant et al. reported the first case treated with eculizumab [15]. Severe delayed hypersensitivity transfusion reaction was defined as acute chest syndrome with worsening hypoxia or acute pulmonary hypertension, stroke or end organ damage [16]. Our patient’s improvement after eculizumab suggests involvement of the complement pathway in this reaction. Establishment of a gold standard therapy in such a cohort requires further study into delayed hypersensitivity transfusion reaction and eculizumab.
CONCLUSION
Hyper-haemolysis is a rare and often fatal type of delayed hypersensitivity transfusion reaction in patients with sickle cell disease. Eculizumab may show promise as the treatment for delayed hypersensitivity transfusion reaction in these patients. Further research should be done regarding the use of this monoclonal antibody as a possible medication of choice in this rare disease process.