ABSTRACT
Multicentric Castleman disease (MCD) represents a group of poorly understood lymphoproliferative disorders related to proinflammatory hypercytokinaemia. In immunocompetent patients the aetiology is still unknown, hence the designation of idiopathic MCD (iMCD). To successfully diagnose iMCD, diagnostic criteria must be fulfilled and a large array of alternative diagnoses excluded. Peripheral neuropathy and nephropathy are relatively common findings in cases associated with POEMS syndrome, but very rarely reported in iMCD.
We present the case of a 64-year-old man with iMCD (HIV- and HHV-8-negative) with nephrotic syndrome and severe motor polyneuropathy. Alternative diagnoses were excluded. The patient was treated with intravenous glucocorticoid followed by rituximab. Complete clinical and laboratory remission was achieved and maintained at the 2-year follow-up.
LEARNING POINTS
- iMCD is a lymphoproliferative disease in immunocompetent patients with no known cause.
- To diagnose iMCD major and minor criteria must be fulfilled, and alternative diagnoses must be excluded.
- Nephrotic syndrome and motor polyneuropathy are rare in iMCD and all alternative diagnoses must be excluded before relating all of these.
KEYWORDS
Multicentric Castleman, nephrotic syndrome, polyneuropathy
INTRODUCTION
Multicentric Castleman disease (MCD) represents a group of poorly understood lymphoproliferative disorders in which proinflammatory hypercytokinaemia plays a crucial pathogenic role. Patients present with multicentric lymphadenopathy with typical histopathologic characteristics, and commonly single or multiple organ dysfunction [1].
In immunosuppressed patients, human herpesvirus 8 (HHV-8) causes MCD. In immunocompetent patients the aetiology remains unknown, hence the designation of idiopathic MCD (iMCD) [2].
The diagnostic criteria for HHV-8-negative/iMCD in immunocompetent patients state that the 2 major criteria (typical histopathological lymph node features and multicentric lymphadenopathy) and at least 2 minor criteria have to be fulfilled, and other diseases that can mimic the clinical and histopathological features of iMCD need to be excluded [2].
CASE DESCRIPTION
We present the case of a 64-year-old man with history of hypertension.
The patient was admitted due to a 4-week history of generalized oedema and hand and feet paraesthesia. Progressive worsening occurred and quadriparesis emerged 2 weeks before admission, initially predominantly in the lower limbs but with gradual involvement of the upper limbs. At admission, muscular strength was graded as 2 out of 5 in all 4 limbs.
Physical examination showed hepatosplenomegaly, generalized oedema and semiology of right-sided pleural effusion. The rest of the examination was unremarkable.
Laboratory tests showed: haemoglobin 15.2 g/dl; normal leucocytes, platelets and blood smear; unremarkable renal, hepatic and thyroid tests (serum creatinine 0.75 mg/dl); erythrocyte sedimentation rate (ESR) was 71 mm/h and C-reactive protein was normal. Serum ferritin was 466 µg/l. Urinalysis revealed nephrotic proteinuria (5.5 g in 24-hour urine collection). Serum albumin was 1.1 g/dl. Serum antibodies related to systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA) were not detected. HIV, HHV-8, hepatitis B and C, EBV and CMV were also excluded.
Lumbar puncture depicted an albuminocytologic dissociation, with no evidence of infection or autoimmune disease. Electromyography suggested chronic inflammatory demyelinating polyneuropathy.
A CT scan revealed multiple mediastinal, coeliac and mesenteric lymphadenopathy, moderate splenomegaly, moderate ascites and right pleural effusion, which was drained and its analysis revealed a transudate with negative cultural and histological examination findings.
A surgical excisional biopsy of an intra-abdominal adenopathy was performed. Its histopathological analysis was suggestive of Castleman disease, hyaline vascular type, with no evidence of lymphoma (Fig. 1).
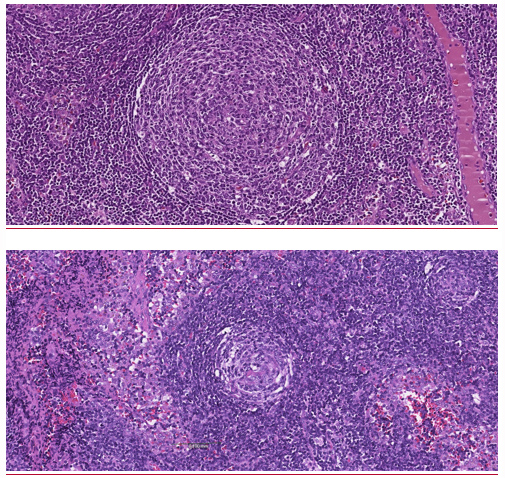
Figure 1 (click to enlarge)
Figure 1. Abdominal lymph node biopsy – HE: sections show regressive follicles surrounded by sheets of plasma cells. Perifollicular blasts were absent
HHV-8 infection was excluded through immunohistochemistry (LANA-1) on the lymph node and through measurement of the serum HHV-8 viral load by polymerase chain reaction. Peripheral blood immunophenotyping was unremarkable.
A bone marrow aspirate and bone marrow biopsy depicted reactive bone marrow.
A renal biopsy was performed and showed a membranous nephropathy, with no traces of amyloid or immune complexes. The search for amyloid on abdominal fat biopsy was negative. A monoclonal gammopathy was excluded. Serum cryoglobulins and antineutrophil cytoplasmic autoantibodies were not detected.
Serum levels of interleukin-6 were elevated = 4.5 pg/ml (normal range 0–3.4 pg/ml).
The patient was started on intravenous glucocorticoid 1 mg/kg per day (80 mg) for 4 weeks, later tapered. The patient was discharged on the 32nd day on oral prednisolone. At that time he presented grade 5 muscular strength in all limbs. He was referred to a specialized centre and was started on rituximab 700 mg (375 mg/m2) once a week for 4 weeks. Prednisolone was then tapered for 10 months, guided by the progressive reduction of proteinuria.
On follow-up at the end of treatment, there was complete clinical and laboratory remission (without peripheral nephropathy, proteinuria, lymphadenopathies, organomegaly, pleural effusion or ascites). Currently there is no evidence of relapse, more than 2 years after stopping rituximab.
iMCD accounts for one-third to one-half of all cases of MCD [2].
Typically, the disease is associated with interleukin-6, the measurement of which might contribute to diagnosis and treatment [5].
Our patient was diagnosed with iMCD according to the diagnostic criteria (Table 1) [2]. Both major criteria were met – multicentric lymphadenopathy and characteristic histopathological lymph node features – as were multiple minor criteria – hepatosplenomegaly, pleural effusion/generalized oedema/ascites, elevated ESR and hypoalbuminaemia [2].
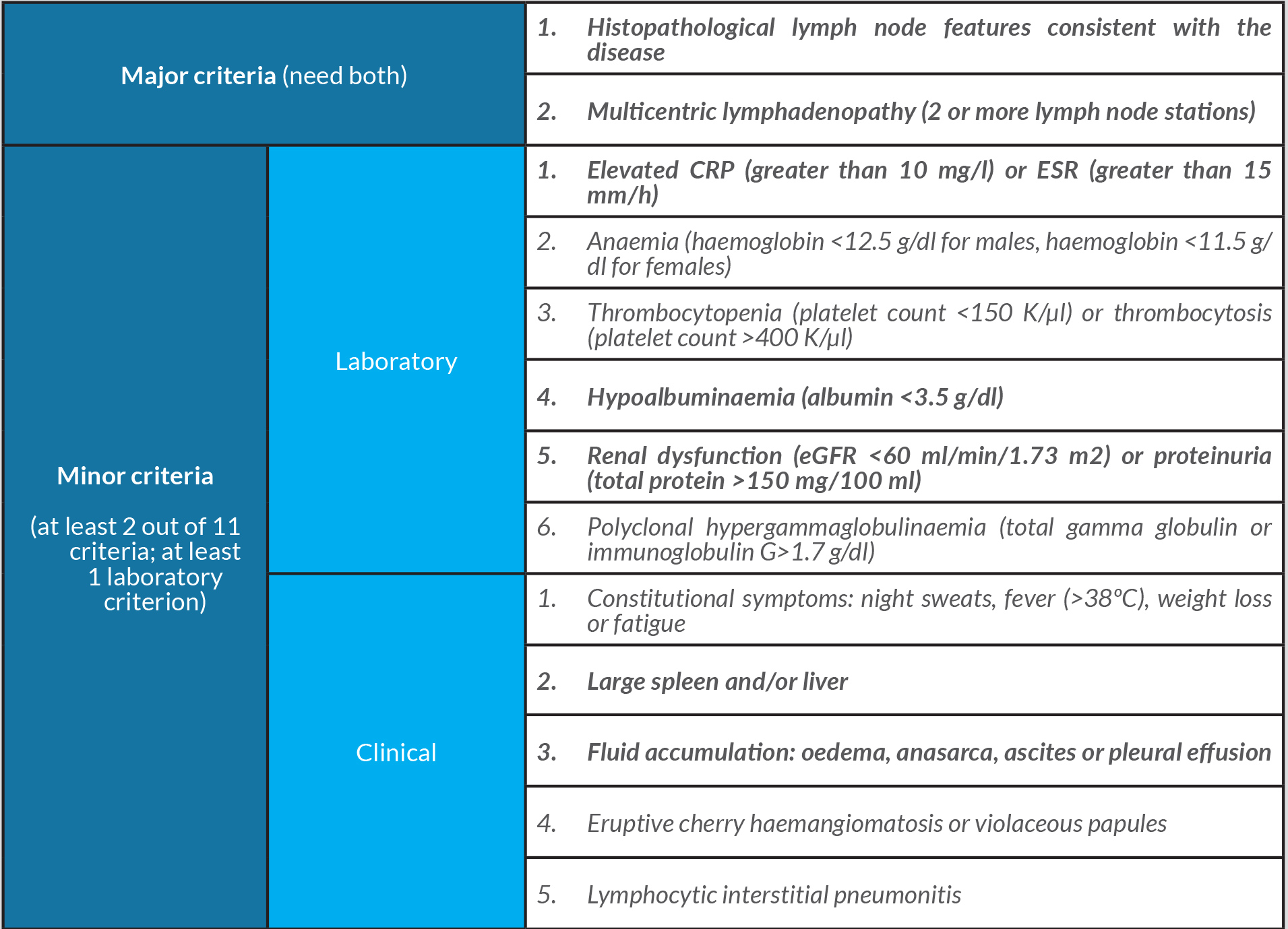
Table 1 (click to enlarge)
Table 1. Diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Highlighted criteria were fulfilled by our patient
The histopathological changes and clinical abnormalities in MCD are nonspecific and may be present in several malignant, infectious and autoimmune conditions. Therefore, these diseases had to be excluded before a diagnosis of iMCD was made (Table 2) [2], namely HHV-8, HIV, EBV and CMV infections, as well as SLE, RA, toxoplasmosis, active tuberculosis and adult-onset Still’s disease. Other lymphoproliferative diseases were excluded through the histopathological analysis of the surgically removed lymphadenopathy. Multiple myeloma and POEMS syndrome were excluded by the absence of monoclonal gammopathy or characteristic changes in bone marrow aspirate and biopsy.
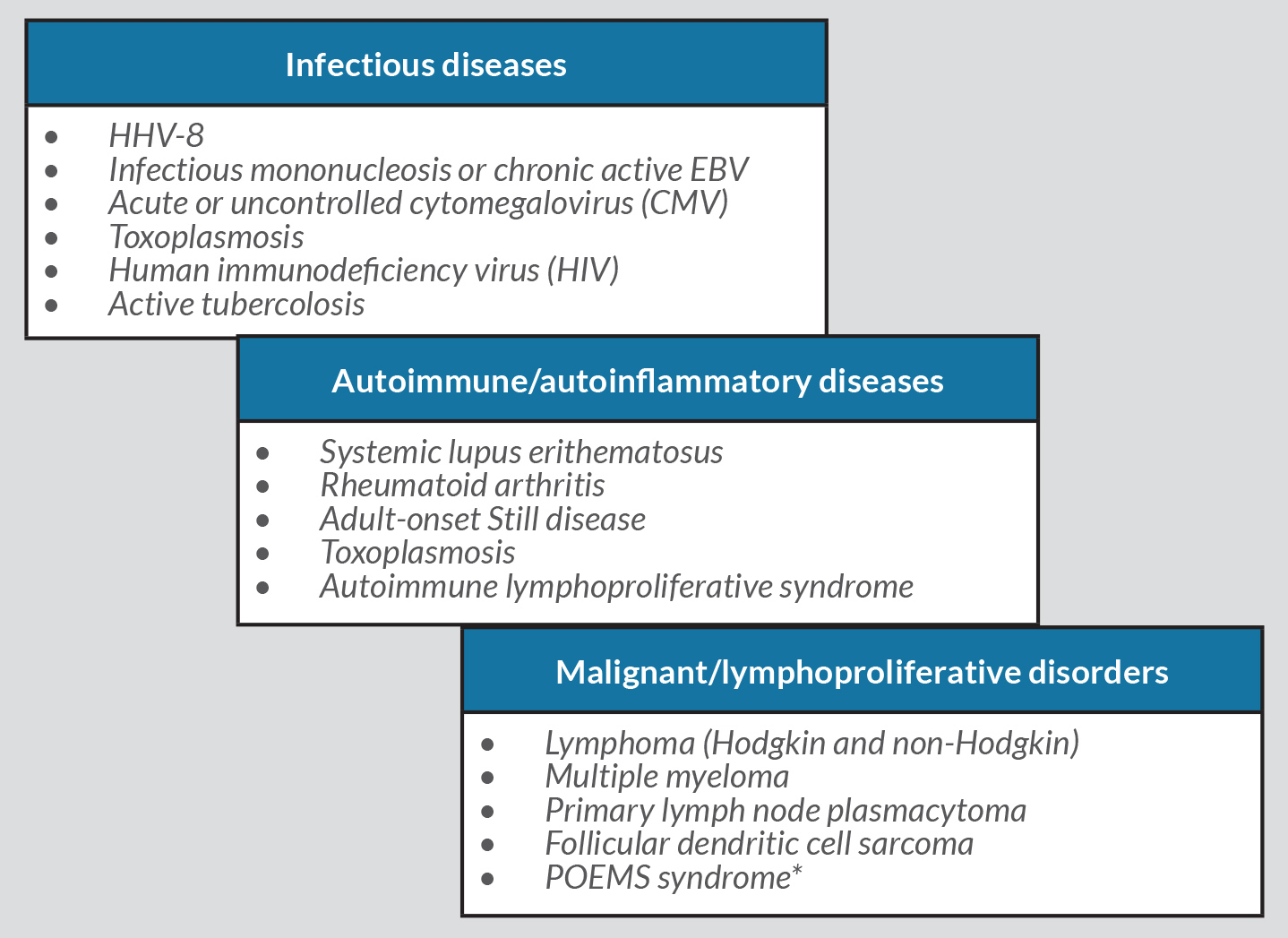
Table 2 (click to enlarge)
Table 2. Differential diagnosis of idiopathic multicentric Castleman disease. *Castleman disease is a major criterion for POEMS syndrome (Dispenzieri, 2017 [6])
Renal disease and polyneuropathy are uncommon manifestations of iMCD, and are more often associated with POEMS syndrome.
Renal disease in MCD is reported in up to 50% of cases, even though renal biopsies are rarely obtained [4]. Its presentation is very variable but it rarely presents with nephrotic syndrome [7]. In HIV-negative patients with Castleman disease and nephrotic syndrome, the spectrum of histological patterns is very heterogeneous and often severe [7, 8]. Our patient’s renal biopsy revealed a membranous nephropathy, which is consistent with previously reported renal manifestations of iMCD.
Castleman disease is associated with peripheral neuropathy in less than a third of cases. It more frequently occurs in cases associated with POEMS syndrome, in which is more severe, painful and with sensory and motor involvement [3, 9]. Polyneuropathy in Castleman disease without POEMS syndrome is predominantly mild, often purely sensory, painless and rarely with motor dysfunction [9]. The motor involvement, especially with the severity presented in this case, is very rare.
A large variety of treatments have been used in iMCD. Rituximab has been shown to be effective in treating HIV-positive and iMCD patients, as well as other systemic diseases with renal involvement, with a favourable safety profile [5, 10].
Even though nephrotic syndrome and severe polyneuropathy are rare in iMCD, we assume a relationship between them since all alternative diagnoses were excluded, and all clinical and laboratory findings regressed completely after treatment, without recurrence approximately 2 years after.
Hence, we present a case in which 2 very rare complications of a very rare syndrome itself were diagnosed, intending to highlight the extremely heterogeneous clinical presentation of iMCD, which can lead to frequent errors in diagnosis with the consequent negative impact on patient outcome.