ABSTRACT
We report the case of a 62-year-old male patient fully vaccinated for COVID-19, admitted to our emergency room for persistent fever associated with exertional dyspnoea, skin lesions, diffuse myalgias and arthralgias not responsive to broad-spectrum antibiotic and antiviral therapy, who developed a rapidly progressive refractory to treatment interstitial lung disease due to anti-melanoma differentiation-associated gene 5 (MDA5) antibodies, that required mechanical ventilation and ECMO. Here, we highlight the importance of always considering alternative diagnoses, i.e. viral and autoimmune diseases, including anti-MDA5 antibody screening, when dealing with patients with a skin rash, seronegative polyarthralgias and interstitial pneumonia, or acute respiratory distress syndrome of unknown origin.
LEARNING POINTS
- MDA5-associated dermatomyositis is a rare systemic syndrome associated with rapidly progressive and treatment-refractory interstitial lung disease.
- The anti-MDA5 antibody is the key biomarker for the diagnosis.
- Early diagnosis is crucial to promptly start aggressive immunosuppressive therapy with the aims of improving prognosis and reducing mortality.
KEYWORDS
MDA5-associated dermatomyositis, MDA5-associated interstitial lung disease, interstitial pneumonia, dyspnoea
CASE DESCRIPTION
A 62-year-old male Italian non-smoker affected by hypertension treated with an ACE inhibitor and a sartan, and fully vaccinated for COVID-19, presented to our emergency room complaining of fever with chills, fatigue, generalized myalgias and arthralgias and exertional dyspnoea over the previous 3 weeks. A chest x-ray prescribed by his general practitioner showed diffuse reticular opacity as an expression of interstitial pneumonia. He was initially treated as having pneumonia, but he failed to respond to 3 days of levofloxacin 500 mg, ceftibuten 400 mg and oseltamivir 75 mg daily, and low molecular weight heparin at a prophylactic dose. He denied weight loss, allergies or exposure to allergens and communicable diseases, and recent travel history. He worked as a policeman. He could not remember chicken pox during childhood. His wife and relatives were all in good clinical condition.
At admission he had a skin rash on his face, chest, hands and legs which evolved through maculopapular, vesicular and crusting phases over 3 weeks (Fig. 1). He was tachypnoeic (22 breaths/minute) with fever (38°C) and oxygen saturation reduced to 93% on room air, 88–89% for minor efforts, including speaking. Blood pressure and cardiac rate were normal, respectively 120/70 mmHg and 88 beats/minute. Musculoskeletal examination did not show any muscle weakness, either proximally or distally, with normal reflexes. Chest examination revealed bibasal crackles. Arterial blood gas analysis confirmed a mild type 1 respiratory failure (PaO2/FiO2 290 mmHg) with pH 7.46, pO2 61, pCO2 35, HCO3 26 and normal lactates, treated with low-flow oxygen by nasal prongs (2 l/min). ECG showed sinus rhythm. Blood tests demonstrated lymphocytopenia, increased erythrocyte sedimentation rate, C-reactive protein (CRP), creatinine, lactate dehydrogenase (LDH), transaminases and creatine kinase (CK), as reported in Table 1.
Troponin I and coagulation time were normal. An RT-PCR nasopharyngeal swab excluded a SARS-CoV-2 infection. Serology tests for COVID-19 were positive for IgG and negative for IgM. LUS revealed a diffuse interstitial syndrome with focal B-lines, mainly distributed in the lower posterior lung fields, with irregular pleural thickening and small subpleural consolidations, in the absence of pleural effusion (Fig. 2). A high-resolution CT (HRCT) chest scan confirmed the diagnosis of interstitial pneumonia with diffuse ground-glass opacities and bilateral basal small consolidations in the absence of lymphadenopathies (Fig. 3).
The patient was admitted to our emergency ward with a diagnosis of interstitial pneumonia highly suspicious for varicella-zoster virus (VZV). Rash distribution was suggestive of varicella disease with erythematous macules evolving to small papules and then crusts, mainly distributed in the trunk with centrifugal spread. Acyclovir was promptly started. After 3 days, normalization of CRP and the white blood count and a significant reduction in CK levels were observed (Table 1, Day 3), despite the development of a progressive acute respiratory failure treated with high-flow nasal cannula. Laboratory findings showed elevated serum levels of myoglobin, increased transaminases, LDH and serum ferritin with normal transferrin saturation (26%) (Table 1, Day 5).
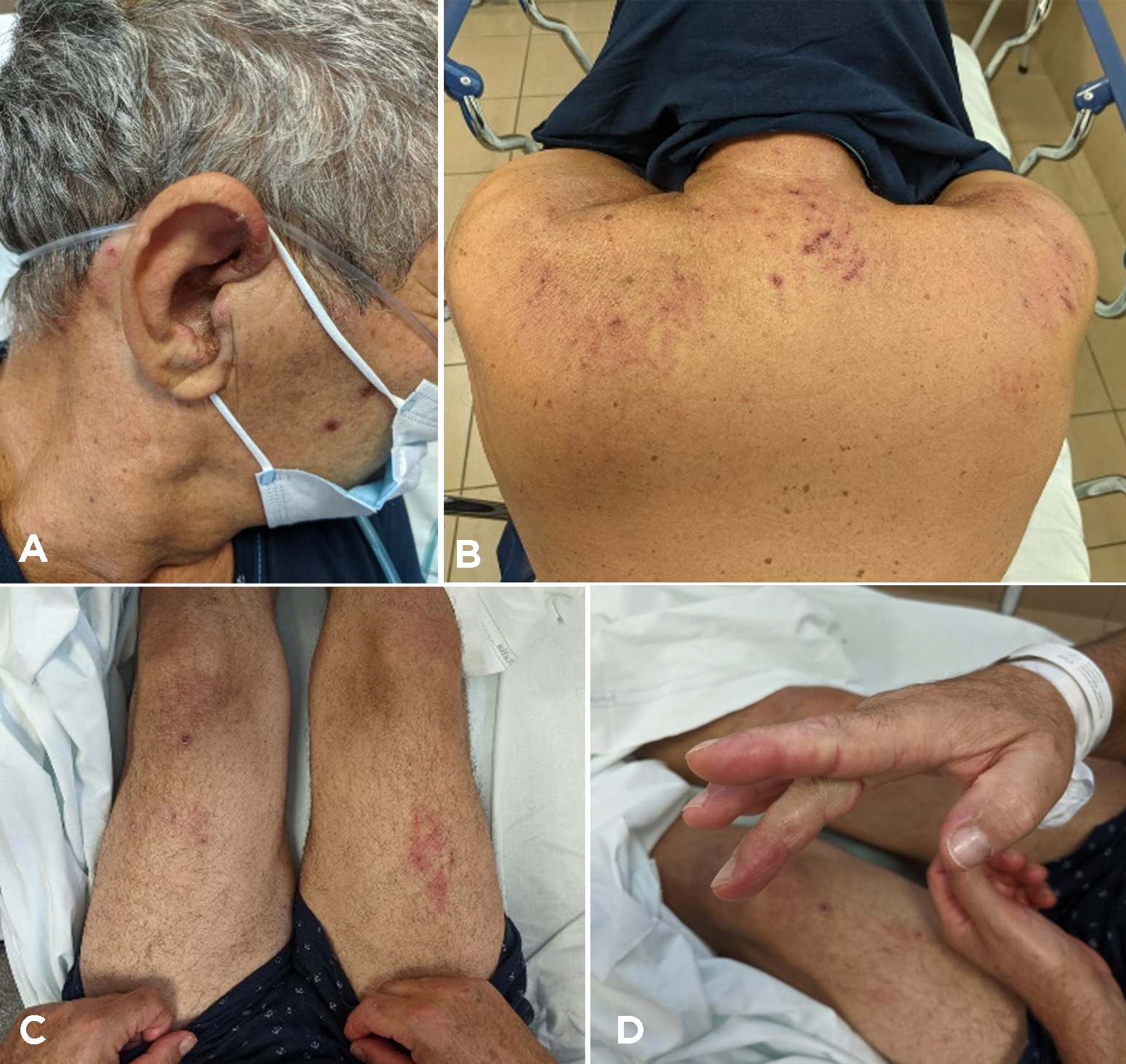
Figure 1 (click to enlarge)
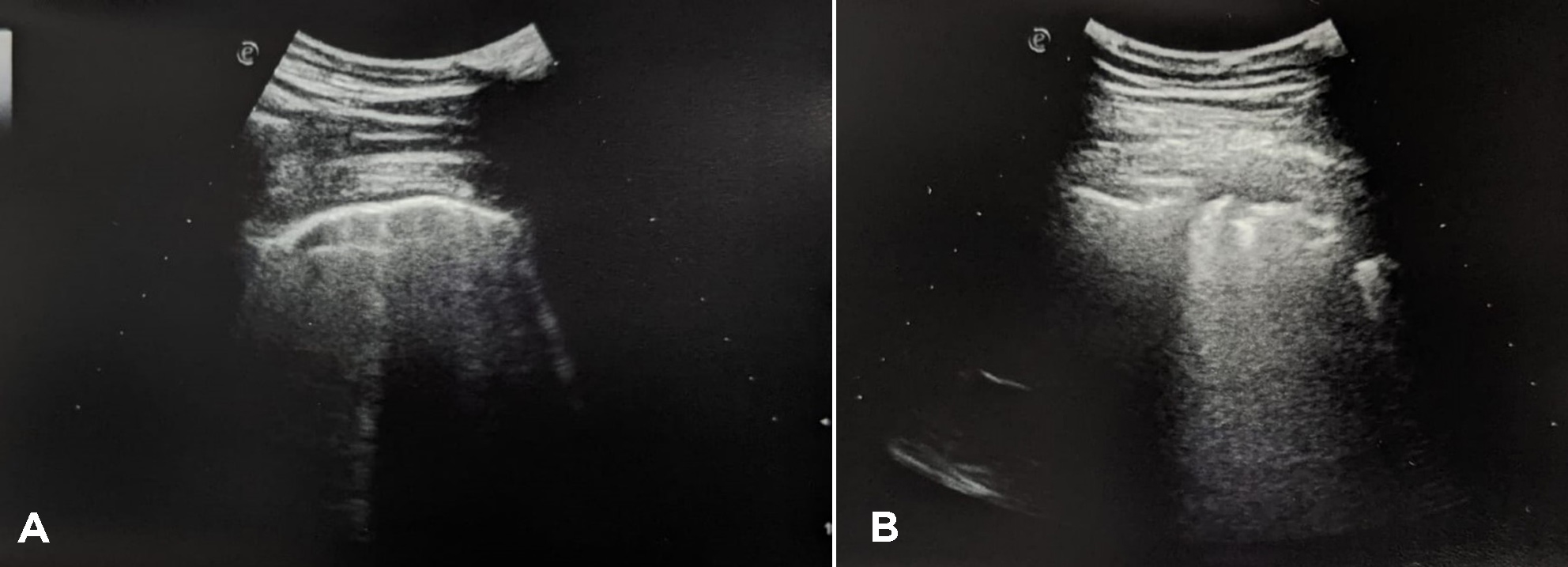
Figure 2 (click to enlarge)
Figure 1. Skin lesions at admission (A–D)
Figure 2. LUS showing a B-line pattern (A) with pleural irregularities and small subpleural consolidations (B) in the lower fields
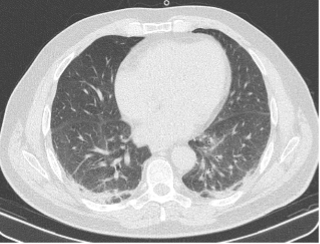
Figure 3 (click to enlarge)
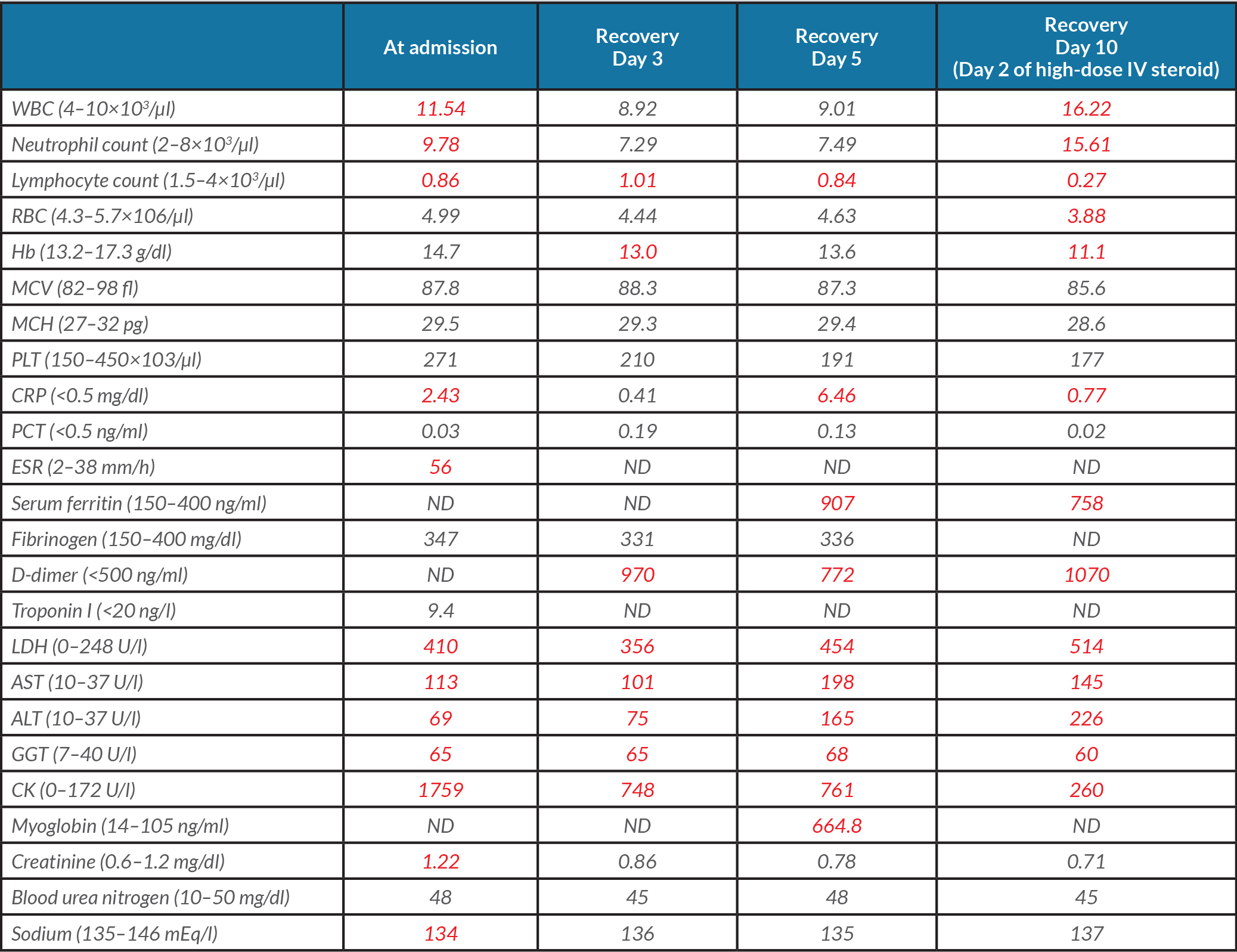
Table 1 (click to enlarge)
Figure 3. HRCT chest scan at admission showing diffuse ground-glass opacities and bilateral basal small consolidations
Table 1. The patient’s laboratory findings at admission and during recovery.
WBC: White blood cell count; RBC: red blood cell; Hb: haemoglobin; MCV: mean cell volume; MCH: mean cell haemoglobin; PLT: platelet; CRP: C-reactive protein; PCT: procalcitonin; ESR: erythrocyte sedimentation rate; LDH: lactate dehydrogenase; AST: aspartate transaminase; ALT: alanine transaminase; GGT: gamma-glutamyltransferase; CK: creatine kinase; ND: not determined. Abnormal values are in red
Repeat blood and urine cultures were negative. Serial procalcitonin was always in the normal range. Serology for HIV, HAV, HBV, HCV, EBV, Cytomegalovirus (CMV), parvovirus B19, VZV, HSV-1/2, rubella, measles, Chlamydia, Mycoplasma pneumoniae, Legionella, pneumococcus, Borrelia, Leptospira, Trichinella, Treponema pallidum, Coxiella excluded an active infection. Urinary antigen testing for pneumococcus and Legionella was negative. The study of lymphocyte subpopulations in peripheral blood showed a normal count and distribution. Protein electrophoresis patterns were normal. IgG, IgA and IgM were in the normal range. An autoimmune panel was requested. ENA, AMA, ASMA, anti-LKM, rheumatoid factor, anti-CCP antibodies, P- and C-ANCA were all negative, except for ANA which was weakly positive (1:320). C3 and C4 levels were normal. Thyroid function was normal with anti-thyroid antibodies absent. Systemic steroid therapy with methylprednisolone (1 mg/kg bid) and empiric broad-spectrum antibiotic therapy with ceftriaxone (2 g) and azithromycin (500 mg) were started.
On Day 5, the patient developed a new skin rash with painful palmar papules, periungual microhaemorrhages and Raynaud's phenomenon, and a severe respiratory failure. Helmet C-PAP with PEEP support of 10 cmH2O was promptly started. A HRCT scan was repeated showing multifocal ground-glass opacities, interlobular septal thickening and increased bibasal consolidations; these findings were consistent with a rapidly progressive interstitial lung disease (RP-ILD) (Fig. 4). A bronchoscopy was performed. Bronchoalveolar lavage testing was positive for Pneumocystis jirovecii, treated with trimethoprim-sulfamethoxazole. An echocardiogram and cardiac MRI were normal. A CT scan of the abdomen did not show any definite evidence of abdominal malignancy.
The combination of RP-ILD, skin rash and muscle involvement was suggestive of dermatomyositis (DM) with lung involvement, as confirmed by the presence of anti-melanoma differentiation-associated gene 5 (MDA5) autoantibodies on the myositis screen. A diagnosis of MDA5-associated DM with RP-ILD was made. The patient was treated with intravenous methylprednisolone 1 g daily with a significant reduction in CK and CRP (Table 1, Day 10), and amelioration of skin rash, despite a rapid deterioration of his respiratory function with the development of acute respiratory distress syndrome (ARDS) 48 hours later (Fig. 5). The patient was intubated and transferred to another hospital specializing in rare diseases and equipped for extracorporeal membrane oxygenation (ECMO) (IRCCS Policlinico San Matteo, Pavia, Lombardia, Italy). ECMO was immediately started at admission to the ICU. The patient developed severe complications due to septic shock for Pneumocystis jirovecii infection, which is a contraindication for an aggressive immunosuppressive therapy. High-dose intravenous immunoglobulin (IVIG) treatment was started, but lung function dramatically worsened due to the development of bilateral pneumothorax and bronchopleural fistula on mechanical ventilation. As we write, after 2 months of hospitalization, ECMO and mechanical ventilation are still ongoing, the clinical condition of the patient remains severe and the prognosis is poor.
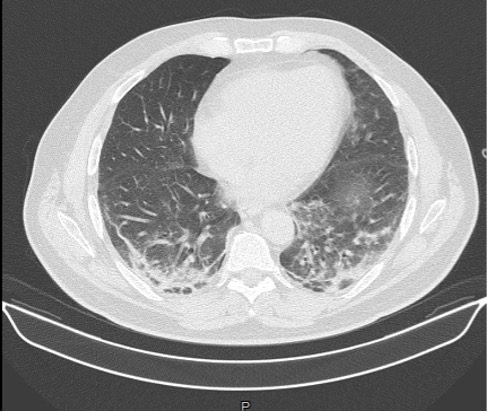
Figure 4 (click to enlarge)
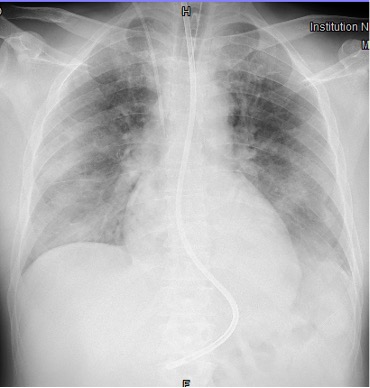
Figure 5 (click to enlarge)
Figure 4. HRCT chest scan showing the worsening of multiple small consolidations and multifocal ground-glass opacities in both pulmonary lobes
Figure 5. Chest x-ray showing bilateral diffuse lung opacities involving all the pulmonary quadrants as observed in ARDS
DISCUSSION
MDA5-associated DM is a rare idiopathic inflammatory myositis firstly described by Sato et al. in 2009 [1] with distinctive mucocutaneous features and a lower incidence of myositis (hypo- or amyopathic DM) but, more importantly, an elevated risk of rapidly progressive and treatment-refractory ILD (RP-ILD) with a potentially fatal course [2]. A limited number of case reports and case series have been reported up to now. As reported in a recent case study by Allenbach et al. [3], MDA5-associated DM must be considered a systemic syndrome, the so-called “anti-MDA5+ syndrome”, rather than a musculocutaneous disease. The authors identified and described 3 clusters of anti-MDA5+ patients with different clinical features and prognoses: the “RP-ILD cluster” with severe lung involvement and a poor prognosis with a very high mortality rate, the “rheumatoid cluster” with anti-MDA5+ rheumatic DM and a good prognosis and the “vasculopathic cluster” with severe skin vasculopathy and an intermediate prognosis [3]. Interestingly, our patient belongs to the “vasculopathic cluster”: he had signs of skin vasculopathy with Raynaud’s phenomenon, skin ulcers, digital necrosis and increased CK levels. As reported by the authors [3], among patients within this cluster, a small percentage of 22.7% develop RP-ILD. Unfortunately, the diagnosis is often overlooked because the acute presentation with predominant lung involvement may mimic other causes of interstitial pneumonia, including COVID-19 [4] or other viral infections, as occurred in our case. ILD is the most important systemic complication and the major determinant of survival, being often rapidly progressive and associated with a poor prognosis. The prevalence of ILD is higher than observed in typical DM, ranging from 42 to 100% depending on ethnicity [5]. It is pivotal to diagnose MDA5-associated DM early in order to promptly start aggressive immunosuppressive therapy to improve prognosis. The most characteristic and striking disease signs are cutaneous ulcers, Gottron papules and painful palmar plaques involving the digital pulp and finger nail folds, suggestive of an underlying vasculopathy [6]. Diagnosis is made by detecting MDA5 antibodies that are DM-specific and appear to be mutually exclusive with other myositis-specific antibodies [3, 7]. Therapy is still largely empiric and should be individualized. There is a dearth of clinical studies based on observational studies and case reports evaluating the best line of treatment, but there are no guidelines [4, 8]. Steroid monotherapy and standard immunosuppressants, such as methotrexate, azathioprine and cyclosporin A, have shown poor results in RP-ILD with a fatal outcome for pulmonary insufficiency in a few months [9]. Current evidence suggests pulse corticosteroids (15–20 mg/kg/dose) for 3–5 days followed by oral corticosteroids (1 mg/kg) along with combination therapy of immunosuppressants for induction [10]. In a multicentre prospective study by Tsuji et al. a combination of steroids, tacrolimus and cyclophosphamide (IV CYC) showed significantly better 6-month survival rates as compared with a historical cohort which underwent step-up immunosuppression (89 vs 33%) [10]. A similar rate of serious infections was recorded in both groups, except for higher CMV reactivation in the former (85 vs 35%). All 3 deaths observed in the combination group were due to worsening ILD. The authors suggested using plasmapheresis for additional effectiveness in intractable disease. In refractory cases, plasma exchange, polymyxin B haemoperfusion and IVIG exhibit a variable response [11, 12]. When the use of calcineurin inhibitors is not feasible, mycophenolate mofetil and rituximab have been used, although unsuccessfully in some case reports [13]. In a retrospective study by Mao et al. ultra-low-dose rituximab (100 mg) may be effective in ILD patients but causing a pronounced and prolonged B-cell depletion with a significant increased rate of opportunistic infections and fatal cases within 2 months of treatment [14]. Despite aggressive treatments with high-dose glucocorticoids in combination with immunosuppressive agents, the 6-month mortality still remains as high as 50% [15]. For this reason, another class of drugs has been recently investigated: the Janus kinase (JAK) inhibitors. Japanese investigators have proposed tofacitinib as a rescue option for patients with high-risk ILD after failure of conventional treatment [16]. More recently, Chen et al. have suggested JAK inhibitor treatment in the early stage of disease before pulmonary function severely decompensates. In their study tofacitinib use led to an improved 6-month survival (100 vs 78%), reduction in ferritin levels and CT scores in patients with ILD [17]. Pirfenidone, an oral antifibrotic agent approved for the treatment of idiopathic pulmonary fibrosis, has been tested in patients with RP-ILD in addition to conventional treatment, showing a significantly higher survival rate only in the subacute (disease duration 3–6 months) ILD patients, but not in the acute (disease duration <3 months) group [18]. Recently, Romero-Bueno et al. have listed 13 recommendations regarding the treatment of RP-ILD using research-based evidence and expert opinion [19]. The authors show that combination of high-dose glucocorticoids and calcineurin antagonists with or without cyclophosphamide is the first-line therapy, but they recommend a tailored therapy based both on the clinical course and the response to treatment. Plasmapheresis, polymyxin B haemoperfusion and/or IVIG may be used as rescue options. ECMO should be considered in life-threatening conditions as a bridge therapy awaiting response to immunosuppressants or to lung transplant. The authors report 8 cases of lung transplant with an overall survival rate of 75% over 7 months to 12 years [19]. In addition, clinicians should remember that opportunistic infection during treatment must be always carefully monitored. Prophylaxis for Pneumocystis jirovecii and CMV reactivation is strongly recommended in the setting of upfront combined immunosuppression.
CONCLUSION
Although MDA5-associated DM is a rare disease, clinicians should always include anti-MDA5 antibody screening when treating patients with skin rash/ulcers, seronegative arthritis and interstitial pneumonia or patients with acute respiratory distress syndrome of unknown origin, particularly if treatment-refractory and rapidly progressive. As reported in our experience, a prompt diagnosis is very challenging, but it remains the key element to avoid fatal complications. In the absence of consensus treatment guidelines, a multidisciplinary approach is mandatory to guide therapy and correctly manage the clinical course.