ABSTRACT
Acquired haemophilia A (AHA) is a rare haemorrhagic disorder caused by the development of autoantibodies inhibiting factor VIII function. It predominantly affects the elderly, who are often burdened with a considerable number of comorbidities, and can result in life-threatening bleeding. The management of AHA consists of two aspects: inhibitor eradication with an immunomodulator and bleed control with a bypassing agent.
Here we present a case of AHA with a high titre inhibitor in a patient with extensive comorbidities and atrial fibrillation in whom inhibitor eradication could not be achieved within a few weeks using corticosteroids alone. Due to coronavirus disease (COVID)-19 restrictions and complications of care, emicizumab offered an effective and convenient therapy, not only sparing the need for continued and intensified inhibitor eradication, but also allowing anticoagulation for stroke prophylaxis.
LEARNING POINTS
- Emicizumab may offer a suitable option for bleeding prophylaxis when inhibitor eradication is not achievable with immunotolerance treatment, especially in the age of the COVID-19 pandemic when the consequences of immunosuppression can be detrimental.
- Bleeding prophylaxis with emicizumab may enable long-term anticoagulation in patients with acquired haemophilia A during inhibitor eradication.
- The prothrombotic risks of emicizumab are not yet sufficiently characterized.
KEYWORDS
Acquired haemophilia A, haemorrhage, emicizumab, anticoagulation, venous thromboembolism, atrial fibrillation
INTRODUCTION
Acquired haemophilia A (AHA) is a rare haemorrhagic disorder caused by the development of autoantibodies inhibiting factor VIII (FVIII) function (a.k.a. ‘inhibitors’). AHA is characterized by severe, often life-threatening bleeding associated with a high mortality rate. AHA predominantly affects the elderly, who are often burdened with a considerable number of comorbidities. In addition, AHA is often associated with malignancy complicating the management of this disorder [1].
To date, the management of AHA consists of two aspects, namely inhibitor eradication and bleed control. Inhibitor eradication involves immunosuppression using immune modulators such as corticosteroids or rituximab (Rituxan®, Genentech, South San Francisco, USA), as well as cytotoxic drugs such as cyclophosphamide (Cytoxan®, Bristol-Meyers Squibb, Deerfield, IL, USA), administered alone or in combination [2]. Inhibitor eradication has been deemed a cornerstone in the management of AHA, since long-term bleed control in the presence of inhibitors against FVIII is difficult to achieve in most patients [2]. The currently approved products to curb bleeding are FVIIa-based bypassing agents (Novo7®, Novo Nordisk, Bagsvaerd, Denmark; FEIBA® and recombinant porcine (rp) FVIII (Obizur®), both from Takeda, Lexington, KY, USA[2]. These clotting factor products have relatively short half-lives (2–10 hours), requiring frequent infusions and indwelling lines making them unsuitable for long-term management in the elderly at home or in long-term care facilities. Hence, rapid inhibitor eradication has been considered critical to enable the discontinuation of clotting factor support [2, 3].
Unfortunately, immunosuppression for inhibitor eradication is not always successful and may take many weeks or even months, with failure rates as high as 30–40% [2, 4]. Moreover, immunosuppression in elderly and frail patients is associated with a considerable number of adverse effects, ranging from 25% to 44%, with infections and sepsis being most common. In fact, infections and sepsis are the most prominent contributors to morbidity and mortality in this population [4].
Most recently though, the approval of emicizumab (Hemlibra®, Genentech, South San Francisco, USA) for congenital haemophilia A with or without inhibitors against FVIII, has opened new avenues for the management of AHA. Emicizumab is a bispecific antibody mimicking FVIII co-factor activity and is administered subcutaneously in weekly, biweekly or monthly intervals depending on dosing. Emicizumab has been shown to be highly effective in controlling bleeding in patients with congenital haemophilia A and inhibitors, as demonstrated by the Haven Clinical Trials Series [5–7]. With respect to AHA, there are several encouraging case reports [8–10], as well as a recent case series demonstrating efficacy [11].
Here we present a case of AHA with a high titre inhibitor in a patient with extensive comorbidities and atrial fibrillation in whom inhibitor eradication could not be achieved within a few weeks using corticosteroids alone. Due to coronavirus disease (COVID)-19 restrictions and complications of care, emicizumab offered an effective and convenient therapy, not only sparing the need for continued and intensified inhibitor eradication, but also allowing anticoagulation for stroke prophylaxis.
CASE DESCRIPTION
A 79-year-old man with a history of multiple comorbidities, including atrial fibrillation, diabetes mellitus and rheumatoid arthritis, was admitted to an outside hospital due to symptomatic anaemia associated with bleeding, thought to be anticoagulation related. The patient was on chronic anticoagulation with apixaban 2.5 mg every 12 hours (Eliquis®, Bristol Myers Squibb, New York, NY, and Pfizer, New York, NY, USA) for stroke prevention. Although information regarding bleeding sites was not clear from the outside medical records, it appeared that most bleeding was musculoskeletal and skin related (ecchymoses). Notably, anaemia and bleeding persisted even after the discontinuation of apixaban, necessitating several hospital re-admissions, involving the transfusion of multiple units of packed red blood cells and fresh frozen plasma, as well as the administration of prothrombin complex concentrates and FVIIa-bypassing agents. The patient had no personal or family history of a bleeding disorder. A new non-resolving traumatic right lower extremity haematoma secondary to a fall prompted a transfer to our institution for further management. At the time of admission, the patient’s haemoglobin levels ranged around 6 g/dl, with mildly elevated white cell and low platelet counts (9,100/μl and 127,000/μl, respectively). The prothrombin time (PT) and the International Normalized Ratio (INR) were normal (12.2 seconds and 1.1, respectively). However, the activated partial thromboplastin time (aPTT) was prolonged to 104 seconds and corrected only partially to 59 seconds with a mixing study (2-hour incubation; normal pooled plasma control = 32.2 seconds). A diagnosis of AHA was suspected and confirmed by FVIII activity (<1%), with an inhibitor titre of 627 Bethesda Units (BU). Computed tomography (CT) of the right leg demonstrated the presence of three prominent intramuscular thigh hematomas, with the largest in the posterior compartment measuring 4.7×5.1×7.4 cm (Fig. 1).
Oral prednisone was started at 1 mg/kg daily for immune suppression, and intravenous rpFVIII was utilized for bleed control based on non-responsiveness to FVIIa-bypassing agents at the outside hospital. The patient received a total of four doses (100–200 U/kg) of rpFVIII within the first 3 days after admission. One-hour post-infusion plasma FVIII activity by one-stage clotting assay was less than anticipated (<10%), suggesting inhibitor cross-reactivity with porcine FVIII (Table 1>. Nevertheless, clinical bleed control with stabilization of haemoglobin was achieved despite low post-infusion levels of FVIII, allowing discontinuation of rpFVIII. The clinical hospital course was complicated by significant hyperglycaemia secondary to high-dose steroid therapy in the setting of diabetes mellitus, which required the initiation of insulin therapy. The patient was discharged on prednisone 1 mg/kg oral daily, off apixaban and with a stable haemoglobin of 8.2 g/dl.
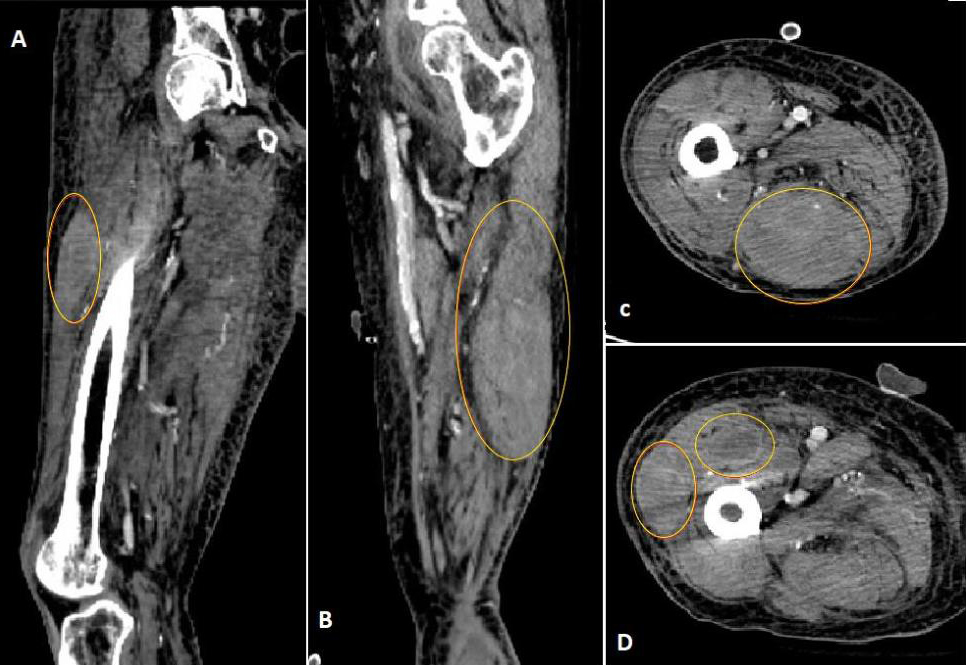
Figure 1 (click to enlarge)
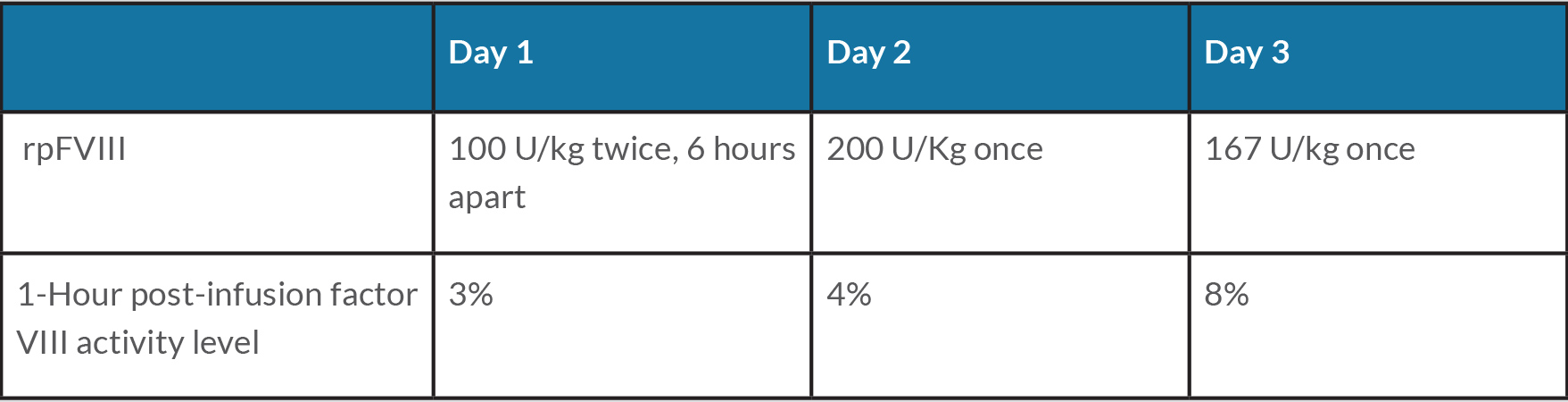
Table 1 (click to enlarge)
Figure 1. Computed tomography of the right lower limb confirmed the presence of three prominent intramuscular haematomas within the right thigh (marked with red circles), with the largest in the posterior compartment measuring 4.7×5.1×7.4 cm. (A) Sagittal section of the right thigh. (B) Coronal section of the right thigh. (C,D) Cross-sections of the right thigh at different levels
Table 1. Treatment schedule and factor VIII level during hospitalization
rp, recombinant porcine
At the patient’s first outpatient evaluation 2 weeks after discharge, the inhibitor titre was still high (749 BU). New areas of ecchymosis encompassing the trunk and extremities were noted and would have required treatment with either rpFVIII, or another trial of more intense FVIIa-based bypassing agents. Due to a complicated living situation and difficult intravenous access, a decision was made to start subcutaneous emicizumab to treat and prevent further bleeding (3 mg/kg subcutaneous weekly for 4 weeks, followed by 3 µg/kg every 2 weeks as per label). If successful, the administration of emicizumab would also abolish the need for immediate continuation and/or escalation of immune suppression, which was deemed increasingly precarious considering the patient’s diminished performance status and risk for COVID-19 infection. Within a few days after the start of emicizumab, bleeding resolved. However, the patient experienced asymptomatic acute non-occlusive proximal left lower extremity deep venous thrombosis (DVT) after several weeks on emicizumab maintenance therapy. FVIII activity levels continued to be undetectable (<1%), as determined by bovine chromogenic assay. The patient underwent inferior vena cava filter placement, followed by a careful risk/benefit analysis with respect to individual bleeding and clotting risks. Recent DVT and a high CHA2DS2-VASc of 6 suggested a high risk for recurrent venous thromboembolism (VTE) and stroke, while continued undetectable FVIII activity levels with a high titre inhibitor level conveyed a high bleeding risk. Considerations included a reduction of the current emicizumab regimen as well as the start of anticoagulation for DVT treatment/prophylaxis and for stroke prophylaxis.
To reconcile competing risks and benefits, a compromise was made by restarting low-dose apixaban (2.5 mg twice daily), while continuing emicizumab maintenance, albeit at a reduced dose and frequency (1.5 mg/kg every 2 weeks). The patient was monitored frequently with physical exams, serial D-dimer measurements, apixaban concentrations (predicted by chromogenic anti-FX assay with drug specific calibrators), FVIII activity and inhibitor titres (bovine chromogenic assays). At the time of this report the patient has been monitored for 9 months without any new bleeding or clotting events.
During this period, his D-dimer level has decreased steadily, with anti-Xa levels within the expected range most of the time (Fig. 2). His haemoglobin also remains stable above 10 g/dl during the follow-up period.
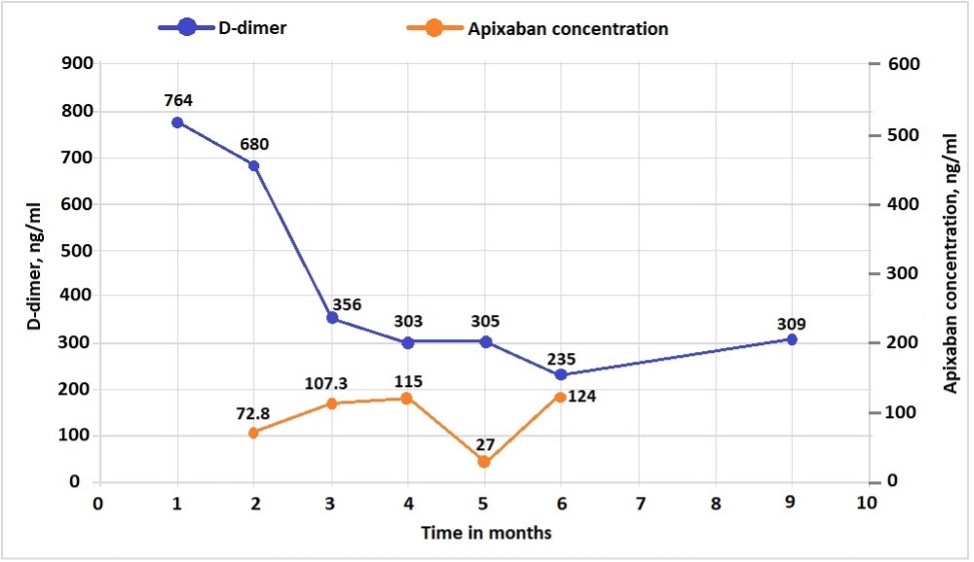
Figure 2 (click to enlarge)
Figure 2. D-dimer and anti-Xa levels after initiation of emicizumab and apixaban therapy.
For D-dimer, in the age-adjusted approach, the cut-off of <500 ng/ml (or 500 µg/l) is retained for patients ≤50 years of age, whereas a threshold of 10 times the patient’s age is used for those >50 years of age [10, 16, 17]. Thus, for a 75-year-old patient, the D-dimer cut-off would be 750 mg/l [6]. Apixaban peak-time concentrations are predicted by the chromogenic anti-Xa method with drug-specific calibrators. The therapeutic concentration of apixaban has not been established. Some studies and data from the manufacturer showed the median peak level for 2.5 mg twice a day dosing to be 123 (69–221) ng/ml [7, 11]e
DISCUSSION
In the treatment of AHA, achieving haemostasis with clotting factor preparations, comprising FVIIa-based bypassing agents or rpFVIII, remains a challenge. Usually, these agents are required to bridge the time until inhibitor eradication is completed using immunosuppression, which was considered the only way to obtain durable haemostasis control in the past [12].
This report highlights an alternative and convenient approach to achieve durable haemostasis with regular subcutaneous emicizumab administration. The haemostatic effect of emicizumab changes a patient’s phenotype from severe to mild haemophilia, but drug levels cannot be assessed by standard coagulation assays. Studies in non-human primates and mice suggest FVIII activity ‘equivalency levels’ of approximately 10–30% in steady-state [13, 14]. Although emicizumab is only approved for congenital haemophilia A with or without inhibitors [5, 6], there is reason to assume effectiveness in AHA, since the mechanism of action (mimicking FVIII co-factor activity [15]) should apply to auto- and alloantibodies alike. In AHA, this attribute would allow the postponement of inhibitor eradication or circumvention of it altogether. This approach may be particularly beneficial for elderly patients with comorbidities, especially in the era of COVID-19. The elderly population are particularly vulnerable to undesired side effects of immunosuppression [2] and are more susceptible to a rapid deterioration in performance status due to delays in making the diagnosis, bleeding complications and hospitalizations.
Several cases and one case series have been published recently describing the efficacy of emicizumab in AHA [8–10]. This case report will add to the literature on several levels. First, early (after the first dose of emicizumab) and durable bleed control (currently 9 months follow-up data) highlight the potential for bleed control and prevention with emicizumab, stressing its potential as a single long-term haemostatic agent in AHA. Downstream, the success regarding durable bleed prevention allowed the discontinuation of immunosuppression. Second, in alignment with cessation of bleeding and immunosuppression, the patient experienced a fast and sustained recovery of overall performance status
Third, this case also highlights that restoration of haemostasis with emicizumab may render patients susceptible to VTE, although risk factors such as age and transient immobility may have contributed. Seven reports have been published of thrombotic events associated with emicizumab use affecting patients with congenital haemophilia A [16–18]: one was arterial, three were venous, and no location was reported for the remaining three events. Two of the venous thromboses were reported in the HAVEN 1 trial and both patients were receiving concomitant treatment with FEIBA at a dose of >100 U/kg/day [16]. There are a few post-marketing reports of thrombotic events in patients with AHA who have been treated with emicizumab [17]. One patient developed pulmonary emboli, one had a thromboembolic stroke, and a third patient died suddenly without detailed clinical information available. Since many patients with AHA are older and may have autoimmune disease and/or cancer, their thrombotic risk may also be higher, which has to be taken into consideration when treating with emicizumab.
Lastly, this case illustrates that anti-coagulation with direct oral anticoagulants is feasible while pursuing bleed protection with emicizumab. Specific to this patient, the need for indefinite anticoagulation was mostly driven by a high stroke risk in the setting of a high CH2DS2 VASc.
While the American Society of Hematology has issued recommendations for anticoagulation therapy for stroke prophylaxis and DVT treatment in patients with congenital haemophilia [18], there are no evidence-based guidelines for AHA. In previous case reports of VTE in AHA [19], treatment of bleeding was prioritized while anticoagulation was suspended with IVC filter insertion. Anticoagulation was either restarted after eradication of inhibitors or not restarted at all [19]. While the concurrent use of emicizumab with anticoagulation has been reported once in the paediatric age group [20], to the best of our knowledge this is the first case to illustrate the concurrent use of emicizumab with anticoagulation in an adult patient with AHA.
CONCLUSION
This report demonstrates that bleed protection with emicizumab might enable (long-term) anticoagulation in patients with AHA during inhibitor eradication, or when inhibitor eradication is not feasible or fails. The prothrombotic risks of emicizumab are not yet sufficiently characterized and may pose a clinical challenge when weighing competing bleeding and clotting risks in individual patients. Therefore, prospective clinical trials to elucidate the effectiveness and risks of emicizumab in AHA are urgently needed.