ABSTRACT
Objective:To report a case of untreated classic 21 hydroxylase (OH) deficiency congenital adrenal hyperplasia (CAH) in a transgender patient resulting in pulmonary embolisms (PEs) and bilateral adrenal masses.
Methods: A 36-year-old male (birth sex: female) presenting with bilateral PEs in the setting of long-standing, untreated classic 21OH CAH was also found to have bilateral adrenal masses (unconfirmed myelolipomas).
Results: Further history revealed a known diagnosis of CAH. The patient had been treated with glucocorticoid and mineralocorticoid replacement in childhood but stopped taking these medications against medical advice. During his hospital admission, he was noted to have elevated 17-hydroxyprogesterone, low cortisol with elevated ACTH levels, and male-level testosterone measurements. CT of the abdomen/pelvis revealed a 23 cm mass in the left renal fossa and a 2.5 cm mass in the right renal fossa consistent with bilateral adrenal myelolipomas. The patient attended follow-up in clinic, but declined any further hormonal treatment as he identified as male and felt further treatment was unnecessary.
Conclusion: This case demonstrated the unique long-term effects of untreated classic CAH due to 21OH deficiency, including bilateral adrenal myelolipoma, adrenal compensation to the point of producing male-level androgens, and possibly PEs. Treatment with hydrocortisone was recommended to suppress ACTH and it was planned that the patient would eventually start on testosterone (although this would have been complicated by his bilateral PEs). Potential aetiologies for the PEs included vascular compression of the renal artery (which could explain the elevated EPO/erythrocytosis contributing to hypercoagulability) or the renal vein by the adrenal mass.
LEARNING POINTS
- Gender dysphoria in patients with congenital adrenal hyperplasia (CAH) is not uncommon.
- Adrenal enlargement can allow untreated CAH patients to compensate.
- Pulmonary embolisms can be a consequence of treating as well as untreated CAH.
KEYWORDS
Gender dysphoria, compensated congenital adrenal hyperplasia, pulmonary embolism
INTRODUCTION
Classic 21 hydroxylase (OH) congenital adrenal hyperplasia (CAH) in XX patients often presents early with virilization. While not common, gender dysphoria is much higher in this group than in the general population. Treatment usually requires hydrocortisone with/without fludrocortisone, although there are documented cases of patients compensating without it. While not typically documented as a complication of CAH, pulmonary emboli are known to be provoked by cancer as well as masses with venoarterial compression. We describe the case of a patient who stopped treatment due to gender dysphoria and the complications arising from that.
CASE DESCRIPTION
A 36-year-old obese male (BMI 52.4) presented to an outlying facility due to acute dyspnoea. He was found to have bilateral submassive pulmonary embolisms (PEs) on CTA of the chest (which also incidentally noted a large solid/fat-containing mass in the left renal fossa) and was started on a continuous heparin infusion. He was then transferred to a tertiary hospital where he was transitioned to an oral anticoagulant in preparation for discharge.
A CT of the abdomen pelvis with/without contrast obtained during hospitalization revealed a left 23 cm mass containing both soft tissue and macroscopic fat elements consistent with adrenal myelolipoma (Figs. 1–3) A right adrenal nodule measuring 2.5 cm was also seen that was in keeping with an adrenal myelolipoma. Further discussion with the patient revealed he had a known diagnosis of CAH at birth. He was previously treated with fludrocortisone/hydrocortisone as well as leuprorelin/somatotropin but stopped treatment sometime in his mid-teens for unclear reasons. Blood tests revealed 17-hydroxyprogesterone of 17,300 ng/dl, AM cortisol of 1.17 µg/dl, DHEA-S of 48.2 µg/dl, total testosterone of 406 ng/dl, and ACTH of 128 pg/ml.
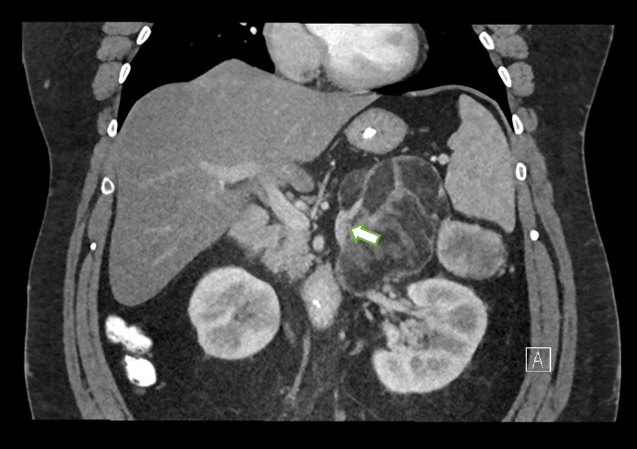
Figure 1 (click to enlarge)
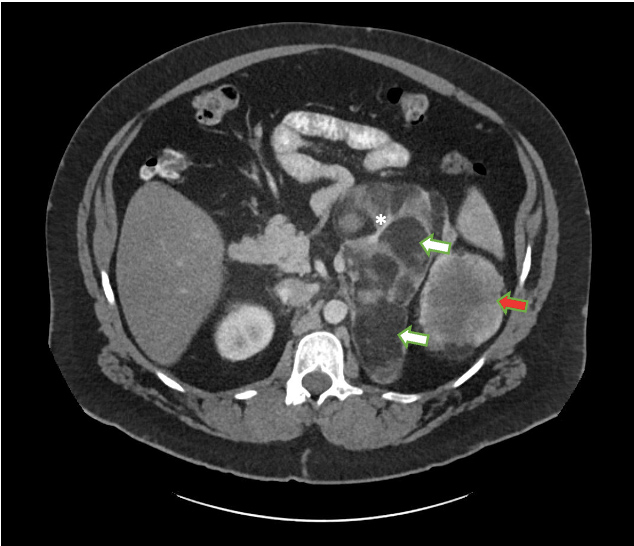
Figure 2 (click to enlarge)
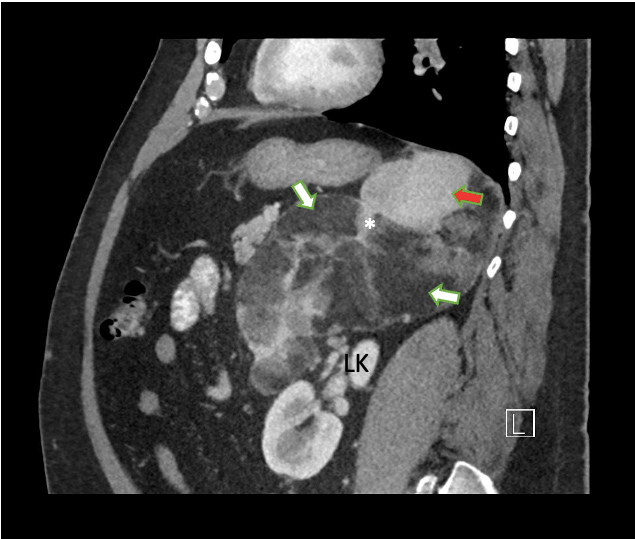
Figure 3 (click to enlarge)
Figure 1. Coronal contrast-enhanced CT images shows an engorged left adrenal vein emanating from the mass, in keeping with an adrenal neoplasm
Figure 2. Large mixed attenuation mass occupying the left suprarenal fossa. Axial contrast-enhanced CT image shows a 23 cm mass containing both fat (white arrows) and enhancing soft tissue elements (red arrows). Several enhancing septations are also noted (*). The mass is separate from the left kidney
Figure 3. Large mixed attenuation mass occupying the left suprarenal fossa. Sagittal contrast-enhanced CT image shows a 23 cm mass containing fat (white arrow). Several enhancing septations are also noted (*). The mass is separate from the left kidney
The option of adrenalectomy was discussed with the patient once he was medically ready. It was also recommended that he resume replacement hydrocortisone and fludrocortisone as he was at risk for adrenal crisis if he decompensated. However, the patient was concerned that this would lower his endogenous testosterone levels and was not interested in the prospect of testosterone replacement. He opted to cancel his future follow-up with endocrinology despite being made aware of the risks.
DISCUSSION
There are documented cases of patients who present initially during infancy with salt wasting due to classic 21OH CAH and subsequently compensate for many years without treatment. This could be due to the normal physiological increase in mineralocorticoid receptors that occurs with age [1]. On the other hand, our patient’s ability to compensate for his lack of glucocorticoid is likely due to increased concentration of androgen precursors with glucocorticoid activity given his significantly low AM cortisol. This type of compensation has also been demonstrated in previous studies [2]. What facilitated compensation for both was also likely related to his massive adrenal enlargement/adrenocortical cell hyperplasia from chronic excess ACTH stimulation that induced glucocorticoid/mineralocorticoid precursor generation. The primary concern regarding CAH treatment for this would be if he would continue to be able to compensate. Patients with 21OH CAH have increased mortality which has been attributed in some part to a failure to compensate in stress situations [3]. Gender dysphoria is not uncommon in 46XX CAH patients raised as female. Declared gender dysphoria is reported in 9% of these patients, and typically first noted in late adolescence/adulthood (similar to our patient). This outcome did not seem to be related to inadequate treatment, null genotype, late diagnosis, severity of virilization, or type of CAH [4]. Female to male transition typically involves testosterone therapy with a goal of 300–1000 ng/dl. This patient’s PEs (as would his baseline erythrocytosis) would typically be a contraindication to testosterone therapy. However, as this specific patient would have been resistant to further management without testosterone therapy, the plan would have been to start testosterone therapy after discussing the risks/benefits with him. It is conceivable his PEs were due to compression of his blood vessels by his large left presumed myelolipoma which would increase the risk of thrombus formation. While this has not been reported as a complication of myelolipomas, it has been seen in patients due to ovarian cysts as well as adrenocortical carcinomas [5]. CAH due to 21OH deficiency itself is associated with an increased frequency of venothromboembolic events, although previous studies on the topic have suggested it may be a consequence of iatrogenic hypercortisolism from excess glucocorticoid replacement (which was not the case with this patient) [6]. Regardless, the exact aetiology of his PEs would have required further coagulopathy studies several months after his initial hospitalization.
CONCLUSION
This patient presents a unique case of untreated compensated classic CAH due to 21OH deficiency which went untreated for many years and led to the development of bilateral adrenal masses, suspected to be myelolipomas. Management for these patients typically involves hydrocortisone/fludrocortisone replacement, which this patient refused as he was asymptomatic. The suggested treatment was complicated as the patient had gender dysphoria (and identified as male) and testosterone therapy would have had to be considered. Also, he presented with bilateral PEs which could have been caused by compression of the renal artery or vein by the large myelolipoma.