ABSTRACT
Castleman disease is an uncommon and heterogenous lymphoproliferative disorder which is classified as unicentric or multicentric depending on the number of lymph nodes involved. Each type has a different clinical presentation, aetiology, treatment and prognosis. We report the case of a young woman who presented with cervical lymphadenopathy and a retroperitoneal mass, and was diagnosed with unicentric Castleman disease and pheochromocytoma. We describe the diagnostic steps, the complications that developed, and the importance of the differential diagnosis in the evaluation of these patients.
LEARNING POINTS
- Castleman disease in an uncommon disease and should be considered in all patients with adenomegalies and systemic symptoms.
- Pheochromocytoma should not be overlooked in the differential diagnosis of a retroperitoneal mass.
- It is important that doctors consider these two diagnoses in patients with adenomegalies and a retroperitoneal mass due to the potential harmful impact of a missed diagnosis.
KEYWORDS
IL-6, adenomegaly, adrenal mass, pheochromocytoma,
CASE DESCRIPTIONS
IWe present the case of a 43-year-old woman with a medical history of untreated hypertension, dyslipidaemia, peripheral venous insufficiency and a former smoking habit.
She was referred for hospital outpatient evaluation due to a cervical lateral lesion which had been growing for 8 years. On presentation, she reported sporadic night sweats. There was no history of anorexia, weight loss, dysphagia, hoarseness, cough, dyspnoea, chest pain or fever. Her familial history was relevant for acute myocardial infarction and cerebrovascular disease.
Physical examination revealed hypertension (blood pressure 166/88 mmHg) and a painless upper-left cervical lymphadenopathy, measuring approximately 30×30 mm. No other lymphadenopathy or organomegaly was detected.
Cervical ultrasound revealed swelling located lateral to the left submandibular gland, and the presence of a hypoechogenic, heterogenous, irregular mass, with lobulated contours measuring approximately 30×16×38 mm with intense vascularization. Cervical computed tomography (CT) was performed with single contrast and confirmed a homogeneous, bulky adenopathy.
Although excision was the most appropriate technique to obtain a correct diagnosis, due to the vascular, friable and pulsatile nature of the lesion and anaesthesia risk, it was deferred and fine needle aspiration biopsy was performed with ultrasound guidance. The postoperative period was complicated with hypertensive acute pulmonary oedema and respiratory failure. The electrocardiogram (ECG) showed sinus rhythm, 100 bpm, inversion of the T wave in aVL and elevation of the J point in V1–V3. Laboratory work-up showed elevation of myoglobin and troponin. The transthoracic echocardiogram (TTE) revealed global hypokinesia, more pronounced in the apex, interventricular septum, and left ventricular inferior and lateral wall, with a left ventricular ejection fraction of 35%. Coronary disease was ruled out by coronary angiography. TTE was repeated and revealed slight hypokinesia of the distal half of all walls and the apex. A diagnosis of Takotsubo cardiomyopathy was considered.
After this complication, right facial palsy developed. Cranioencephalic CT and magnetic resonance imaging (MRI) revealed hyperintense lesions located in the juxtacortical right frontal lobe, the cortico-subcortical left parieto-occipital lobe, the right occipital lobe and in the left cerebellum, compatible with recent ischaemic lesions, which led to the hypothesis of an embolic source. In this context, hypocoagulation with rivaroxaban was initiated.
After this unpredicted complication, the diagnosis of pheochromocytoma was suspected, and 24-hour urine fractionated metanephrines and catecholamines and plasma fractionated metanephrines were collected.
Laboratory evaluation revealed mild thrombocytopenia only (134×109/l). No anaemia, abnormal renal or liver function, thyroid dysfunction, or changes in immunoglobulins or protein electrophoresis were found. Viral serologies were all negative, except for Epstein-Barr virus (EBV) which showed reactive IgG and indeterminate IgM. Human herpesvirus 8 (HHV8) was also negative.
Lymph node histology revealed morphological and immunohistochemical findings compatible with Castleman disease (CD), hyaline-vascular variant (Fig. 1).
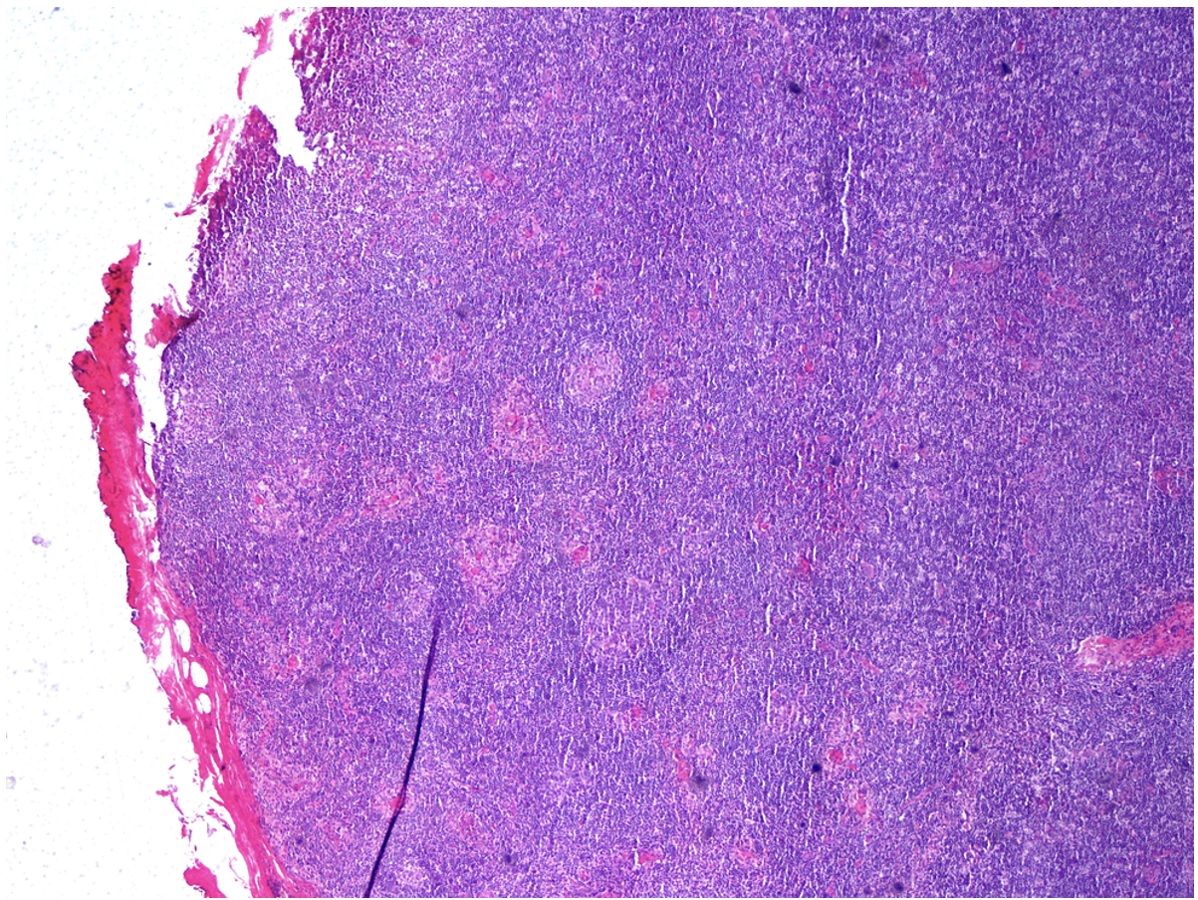
Figure 1 (click to enlarge)
Figure 1. Castleman disease: lymph node histology
Positron emission tomography (PET) with 18-FDG revealed hypermetabolism of the left retromandibular adenopathy, compatible with CD involvement (Fig. 2). There was also very intense radiopharmaceutical uptake in the periphery of a large abdominal mass, suspected of being malignant disease due to the very high metabolic activity. Multiple and diffuse intense radiopharmaceutical uptake was also found in adipose tissue with bilateral distribution in the posterior and lateral cervical, supraclavicular, axillary, mediastinal, juxtacardiac, peripheral, perirenal, mesenteric and inguinal regions (Fig. 3).
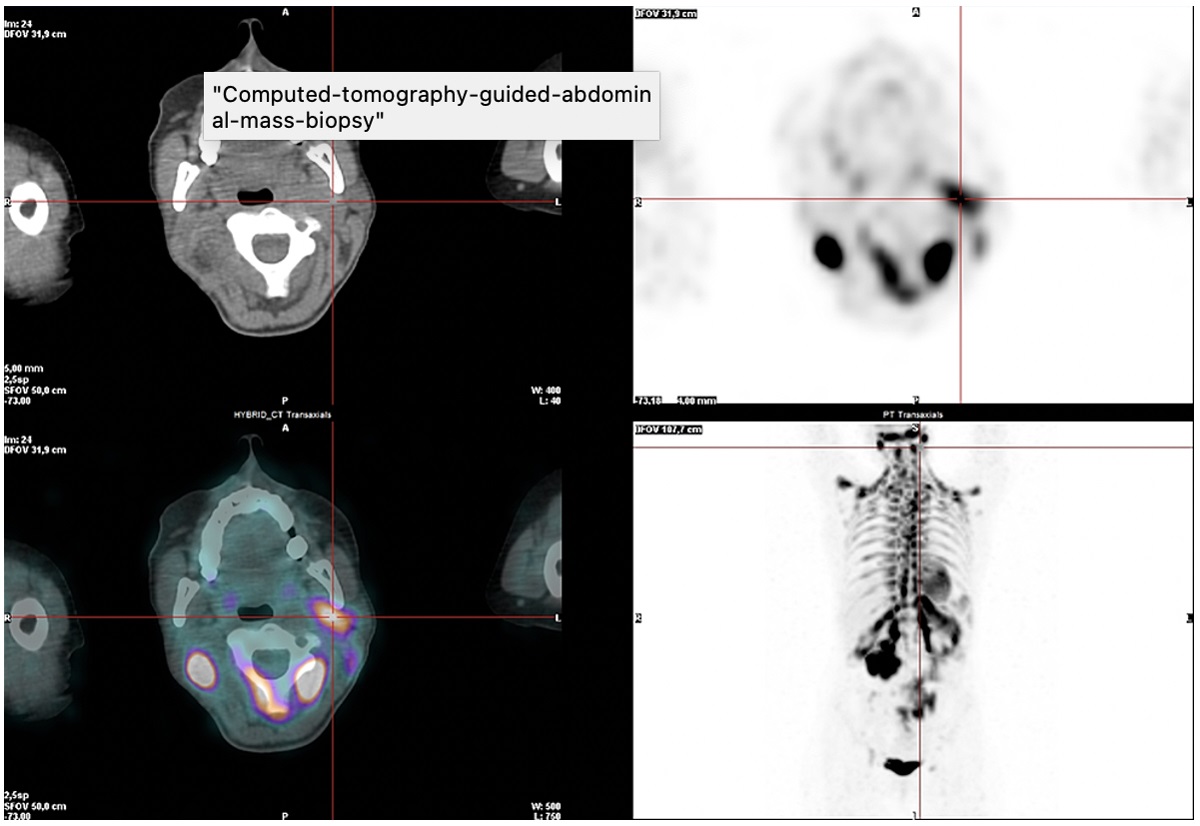
Figure 2 (click to enlarge)
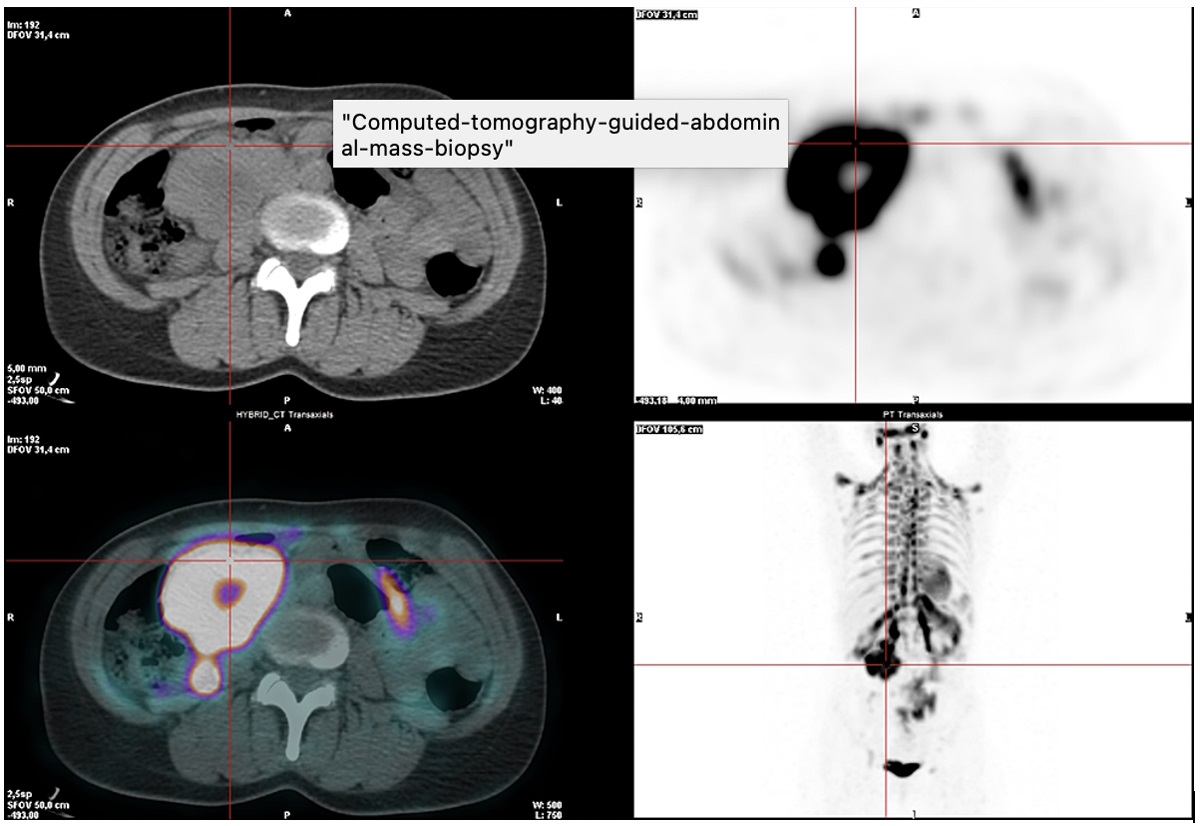
Figure 3 (click to enlarge)
Figure 2. PET scan demonstrating 18-FDG uptake in the left retromandibular adenopathy
Figure 3. PET scan demonstrating 18-FDG uptake on the periphery of a large abdominal mass
Although the metanephrine and catecholamine results were not yet available, it was decided to submit the patient to a CT-guided abdominal mass biopsy for diagnostic clarification and disease stratification. The postoperative period was again complicated with a new episode of hypertensive acute pulmonary oedema that culminated in cardiorespiratory arrest and recovery after two cycles of advanced life support. Due to persistent fever (temperature 39–40°C), severe infection was considered, and empirical antibiotic therapy was initiated.
Pulmonary and abdominal angiography showed no signs of lung thromboembolism. A hypocaptive area about 12 mm long was found in the left ventricular apical region, which could correspond to infarction. Extensive consolidations in the lower lung lobes were identified, probably corresponding to aspiration pneumonia. The patient developed cardiogenic shock refractory to vasopressor support, and started extracorporeal membrane oxygenation (ECMO).
Cranioencephalic CT was performed, showing hypodensities located in the right frontoparietal cortico-subcortical, right polar occipital, left parieto-occipital and left temporomesial lobes, in the left thalamocapsular region, in the left side of the midbrain, the left brain stem, protuberance, and cerebellar hemispheres bilaterally. They eroded the cortical grooves and moulded the IV ventricle, the atrium, and the occipital horn of the left lateral ventricle.
The unfavourable clinical evolution resulted in the patient’s death.
The subsequently reported metanephrine results showed plasma metanephrine 1140 pmol/l (reference range (RR) <456.3), plasma normetanephrine 2477 pmol/l (RR <982.8), urinary metanephrine 2097 pmol/l (RR 264–1729) and urinary normetanephrine 6544.8 pmol/l (RR 480–2424). Histology of the abdominal mass revealed a ‘nested’ neoplasm (of the ‘zell-ballen’ type), consisting of cells with some nuclear polymorphism, sometimes with hyperchromatic nuclei or with pseudo-inclusions. The immunocytochemical study showed neoplastic cells immunoreactive for chromogranin and synaptophysin, while the cytokeratin 8/18 study was negative, confirming the diagnosis of pheochromocytoma but no CD involvement.
DISCUSSION
Castleman disease (CD) was first described in 1954 by Benjamin Castleman. It is an uncommon and heterogeneous disease, defined by the presence of lymph nodes with multiple histopathological features, including atrophic or hyperplastic germinal centres, prominent follicular dendritic cells (FDCs), hypervascularization, polyclonal lymphoproliferation and/or polytypic plasmacytosis [1].
When CD is located in the suprarenal region it is easily mistaken for a suprarenal gland tumour, such as pheochromocytoma. In light of the risk of adverse events during procedures in patients with pheochromocytoma, it is important to consider this entity in the differential diagnosis of a suprarenal mass [2], and wait for all necessary laboratory results to exclude this diagnosis before initiating any procedures.
Unicentric CD (UCD) involves one lymph node or a ganglion region, and in the majority of patients (60%) systemic symptoms are absent. Case series have shown that the median age at presentation is 30–34 years and the disease occurs equally in men and women. In contrast to multicentric CD (MCD), there is no association with human immunodeficiency virus (HIV) or HHV8 infection and no epidemiological risk factors have been identified so far. Patients with UCD have an excellent prognosis with successful treatment [3].
The gold standard for diagnosis is excisional biopsy. Removal of the lymph node is usually curative, resulting in an excellent prognosis and overall survival of 95% [3].
Pheochromocytoma is a catecholamine-secreting tumour rising from the adrenal medulla that commonly presents with paroxysmal hypertension, sweating and tachycardia, as in our patient. It is able to produce a variety of biologically active neuropeptides, hormones other than catecholamines, and inflammatory cytokines, mainly IL-1, IL-6 and TNF-α. It has been suggested that IL-6 produced by the tumour gives rise to pyrexia [4].
Systemic symptoms are present in a small percentage of cases of UCD, and IL-6 appears to be the responsible cytokine, being better characterized in MCD, where these symptoms predominate [3, 5].
One case demonstrating an association between CD and pheochromocytoma has been reported previously. IL-6 appears to be the common link between the two clinical entities. Taking IL-6 as the primary mediator, pheochromocytoma may cause a paraneoplastic syndrome manifesting with fever and marked elevation of inflammatory parameters, unexpected in a patient with unicentric CD, as in the case reported above [4].
Although this is an extremely rare association, and despite the favourable prognosis expected in a patient with unicentric CD, unawareness of the diagnosis of pheochromocytoma, and the need for invasive procedures for definitive diagnosis and staging, contributed to an unfavourable outcome. Pheochromocytoma should be considered in the differential diagnosis of a retroperitoneal mass.