ABSTRACT
Rosai–Dorfman–Destombes disease (RDD) or sinus histiocytosis with massive lymphadenopathy is a rare non-Langerhans cell histiocytosis of unknown cause. The disease often manifests as painless bilateral cervical lymphadenopathy associated with systemic symptoms such as fever and weight loss. Extranodal disease is also frequent and can involve any organ, mostly the skin, nasal cavity, bone, and retro-orbital tissue. Swelling of cartilaginous tissues, such as the helix of the ear or laryngeal structures, may mimic the entity known as relapsing polychondritis. Although spontaneous remission is the most expected evolution, some cases require systemic treatment with prednisone, methotrexate or cytotoxic agents, with variable rates of success. In this respect, since somatic variants in the genes involved in the mitogen-activated protein kinase (MAPK) and extracellular signal-regulated kinases (ERK) pathway have been observed to play a pathogenic role in RDD. Therefore, the use of therapies targeting these pathogenic variants appears to be a reasonable strategy. Here we present the case of a 37-year-old woman with RDD and extensive extranodal involvement that showed a rapid and complete response to the MEK inhibitor cobimetinib.
LEARNING POINTS
- Rosai–Dorfman–Destombes disease (RDD) may mimic the entity known as relapsing polychondritis but should be treated with drug therapy for the underlying disease.
- Mutations in MAPK/ERK pathway components should be determined in RDD with systemic involvement, although testing to determine every somatic mutation responsible for the disease is not available in all healthcare centres.
- MEK inhibitors like cobimetinib could be effective in RDD cases with severe and refractory systemic disease, even if molecular analysis has not been possible.
KEYWORDS
Rosai–Dorfman–Destombes disease, Rosai–Dorfman disease, relapsing polychondritis, MAPK/ERK pathway, cobimetinib
CASE DESCRIPTION
A 37-year-old woman was referred to our hospital due to relapsing polychondritis refractory to corticosteroids. She was an active smoker with no medical history of note, except for an episode of alopecia areata four years before presentation. During the previous year she was evaluated in a private hospital for swelling of the helix of the ear, bilateral persistent episcleritis, and arthralgia of hands, ankles and knees, where the diagnosis of relapsing polychondritis was made. She was treated with high dose corticosteroids, indomethacin and methotrexate, with poor clinical response. Concomitantly, the patient developed asymptomatic and isolated non-scaly erythematous papules affecting both sides of her face (Fig. 1). For the initial diagnosis of granulomatous rosacea, she received topical corticosteroids, topical ivermectin, oral doxycycline and both topical and oral metronidazole, with no clinical response. Indeed, there was progressive extension of the cutaneous lesions, confluent in patches on all sides of the face.
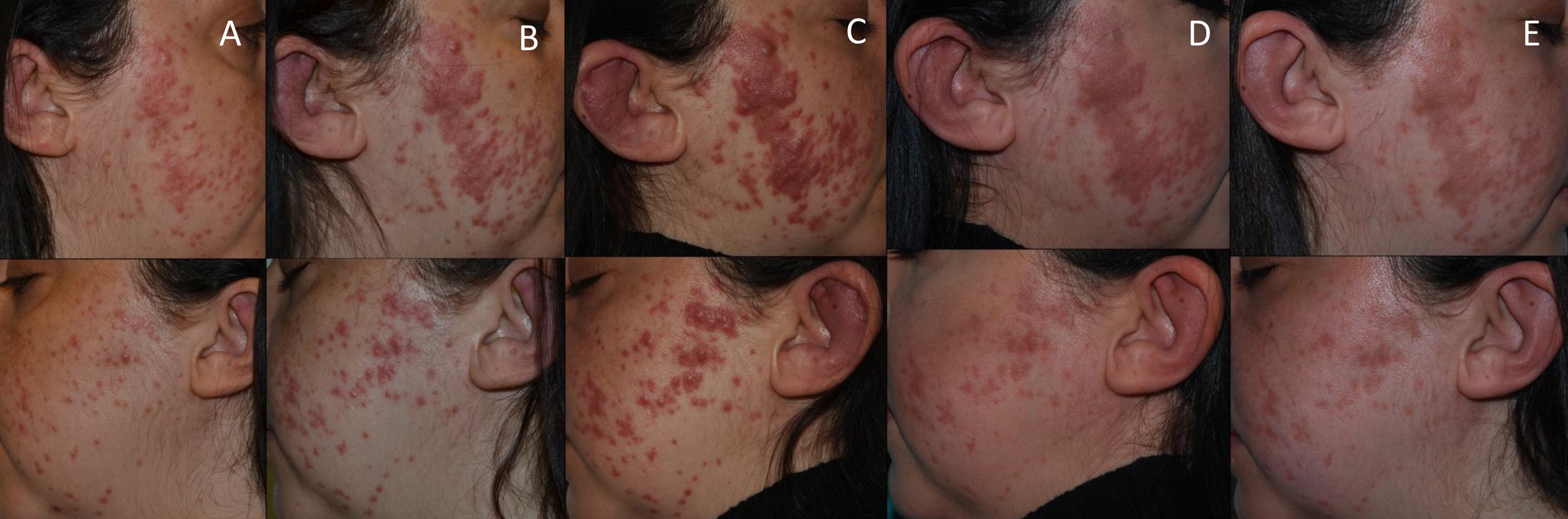
Figure 1 (click to enlarge)
Figure 1. Cutaneous involvement of Rosai–Dorfman–Destombes disease in a female patient who presented with skin and cartilage changes during follow-up. (A) 6 months before cobimetinib administration, (B) 3 months before cobimetinib administration, (C) the day before cobimetinib administration, (D) 1 month after cobimetinib administration, (E) 2 months after cobimetinib administration
Laboratory tests including viral serology and autoimmune tests were negative. A skin biopsy was performed which showed a dense dermal infiltrate of histiocytes in a nodular pattern accompanied by lymphocytes, plasma cells and neutrophils, with evidence of the phenomenon of neutrophil and lymphocyte emperipolesis. Biopsy microbiology tests were negative and immunohistochemical analysis of histiocytes showed positivity for S100 and CD68 and negativity for factor XIIIa and CD1a. These results were compatible with the diagnosis of Rosai–Dorfman–Destombes disease (RDD). In this regard, the B-type Raf kinase V600E mutation (BRAF V600E) was absent in the skin. Furthermore, to assess the extent of RDD, an 18-fluorodeoxyglucose-positron emission tomography/computerised tomography (FDG-PET/CT) was performed, revealing an increased uptake in the auricles of the ear, nasal cartilage, laryngeal and pharyngeal structures, and the aortic wall (Fig. 2A).
Multiple treatments were prescribed during the following 6 months, which included topical tacrolimus, systemic steroids, dapsone, isotretinoin, rifampin and adalimumab, with very limited clinical response. Finally, the patient was admitted to our hospital due to worsening of the outer ear swelling and skin lesions; there was also a marked increase of FDG uptake in the aortic wall and lung parenchyma in a new FDG-PET/CT. Treatment with 250 mg methylprednisolone boluses for 3 consecutive days followed by prednisone 0.5 mg/kg/day were newly administered. However, given the rapid progression of the disease despite multiple drug regimens, the compassionate use of cobimetinib was requested, without prior confirmation of mutations in the MAPK/ERK pathway. In order to avoid adverse events, cobimetinib was administered at low doses (40 mg daily) for the first 21 days of a 28-day cycle. Significantly, shortly after the initiation of cobimetinib, the patient showed improvement of the auricle inflammation and laryngeal symptoms. Accordingly, the corticosteroids were rapidly tapered and 1 month later the patient was no longer receiving corticosteroids. After 1 month of follow-up, there was only residual skin and cartilage involvement (Fig. 1), while repeated FDG-PET/CT showed an almost complete resolution of FDG uptake in all regions previously affected (Fig. 2B). Unfortunately, the patient experienced mild to moderate diarrhoea with no other associated manifestations. Infection or other causes of the diarrhoea were ruled out. Given the persistence of the gastrointestinal symptoms 3 months after treatment initiation, we decided to withdraw cobimetinib, with resolution of the diarrhoea. As RDD typically presents with progressive subacute manifestations, a watchful waiting strategy was adopted. New cobimetinib cycles will be considered should serious RDD manifestations reappear. RDD remains in remission 3 months after the cobimetinib withdrawal.
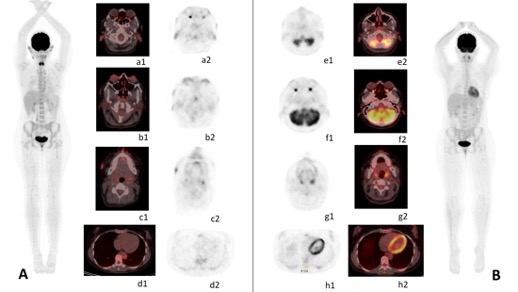
Figure 2 (click to enlarge)
Figure 2. PET/CT changes during follow-up. (2A) Maximum-intensity projection of pre-treatment PET/CT shows multiple hypermetabolic regions in the body. Fused and PET images are shown. These lesions are located in both auricles of the ear (a1-2), laryngeal and pharyngeal structures, both episclera (b1-2), with facial cutaneous (c1-2), aortic wall and periaortic thickening (d1-2). (2B) Maximum-intensity projection of post-treatment PET/CT (after 1 month) shows the complete resolution of FDG uptake in all regions affected in pre-treatment with a persistence of some residual lesions with very slight FDG uptake. Images show the disappearance of FDG uptake in both episclera (f1-2), aortic wall and periaortic thickening (h1-2). Residual lesions are observed in both ear auricles (e1-2) and facial cutaneous localisation (g1-2)
DISCUSSION
RDD or sinus histiocytosis with massive lymphadenopathy is a rare non-Langerhans cell histiocytosis of unknown cause. The disease often manifests as painless bilateral cervical lymphadenopathy associated with systemic symptoms such as fever and weight loss. Extranodal disease is also frequent and can involve any organ, mostly the skin, nasal cavity, bone, and retro-orbital tissue [1]. Skin involvement is a common manifestation of RDD. Typical skin lesions can appear as non-scaly nodules, plaques or papules, which can affect any location on the skin surface. Swelling of cartilaginous structures, such as the helix of the ear, nasal cartilage or larynx, mimicking relapsing polychondritis has rarely been described [2]. A review of the literature found 3 cases of RDD presenting with unusual swelling of the pinnae region of the helix of the ear [2] but to the best of our knowledge there are no previously published cases with biopsy-proven relapsing polychondritis associated with histiocytosis (Table 1).
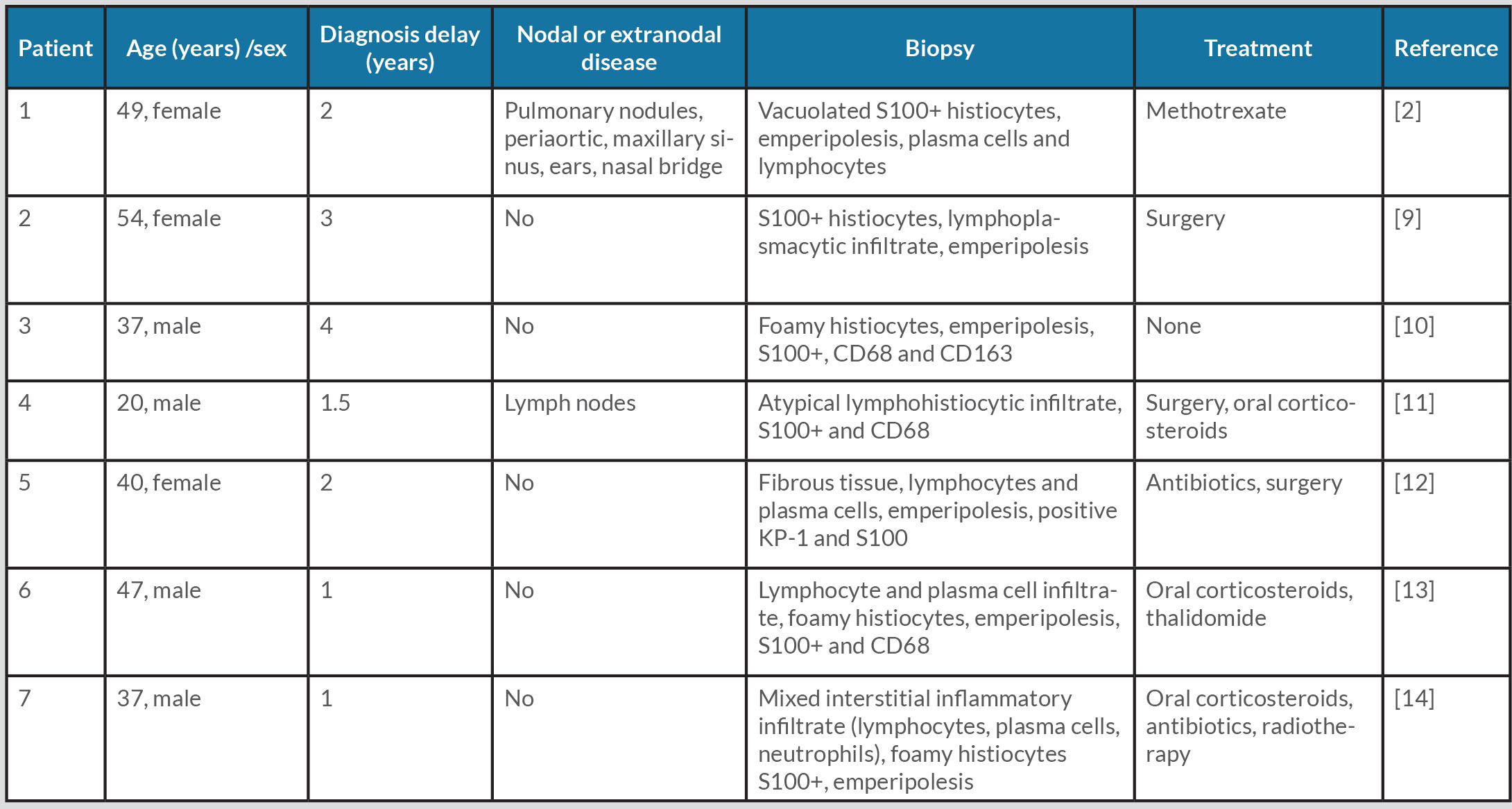
Table 1 (click to enlarge)
Table 1. Reported Rosai–Dorfman–Destombes disease in patients with pinna of the ear involvement
Definite diagnosis is achieved by histopathologic study of involved tissues, showing infiltrates of large histiocytes with emperipolesis (trafficking of intact leukocytes or other cells through the cytoplasm of histiocytes) and an immunohistochemical study showing positivity for S100 and CD68 and negativity for CD1a and langerin [3]. The differential diagnoses of RDD can include IgG4-related disease as well as Langerhans and other non-Langerhans cell histiocytosis. Furthermore, RDD can coexist with immune-mediated diseases in approximately 10% of cases [1] or be associated with haematological neoplasms.
Up to 50% of cases of RDD have spontaneous remission or can be cured with local excision of adenopathies. Systemic treatment is indicated in bulky or symptomatic disease; however, corticosteroids and cytotoxic agents show variable response rates.
Interestingly, promising results have been observed with thalidomide, lenalidomide, and tyrosine kinase inhibitors [4]. Cobimetinib is a selective MEK inhibitor that has been successfully approved in combination with vemurafenib to treat BRAF-mutated melanoma. The MAPK pathway appears to be constitutively activated in metastatic cutaneous melanomas due to a mutation in BRAF V600E, and concomitant inhibition of MEK results in a greater tumour response. Mutations in MAPK/ERK pathway components, such as BRAF, KRAS, NRAS, or MAP2K1, have also been described in some RDD with systemic involvement [1,5]. Indeed, guidelines recommend the MAPK/ERK pathway (KRAS, NRAS, HRAS, ARAF, BRAF, MAP2K1) mutation assessment in severe or refractory cases of RDD [1]. The BRAF V600E test is already available in most healthcare centres, but it is not always possible to evaluate other MAPK/ERK pathway mutations. MEK inhibition is considered a useful strategy in BRAF-wild-type histiocytosis [4] and cobimetinib has proven to be effective in RDD cases with refractory systemic disease [6–8]. Due to the severity and refractoriness of the manifestations experienced by our patient, and even without prior confirmation of the presence of mutations in this pathway, we decided to empirically start cobimetinib at lower dosage (40 mg daily), with an immediate successful response.
In summary, we describe a case of RDD with systemic involvement and severe cartilage inflammation mimicking relapsing polychondritis, refractory to corticosteroids and multiple drug regimens, in which cobimetinib achieved a rapid and complete remission. Further data are needed in order to recommend its routine use in systemic histiocytosis, but it may be an effective therapeutic option in patients with severe disease or refractory to the usual therapies.