ABSTRACT
Budd-Chiari syndrome (BCS) results from an occlusion of the hepatic venous flow which in turn leads to portal hypertension causing ascites and other signs of liver dysfunction. Here, we present the case of a 43-year-old woman with recurrent ascites who was found to have BCS secondary to an inferior vena cava thrombosis extending into the hepatic veins. Although she had a normal platelet count on admission, additional laboratory investigations revealed an MPL mutation. She was discharged on anticoagulation with apixaban and later found to have thrombocytosis on repeat blood work, confirming the diagnosis of essential thrombocytosis, following which she was started on myelosuppressive therapy with hydroxyurea.
LEARNING POINTS
- Complete work-up to evaluate for myeloproliferative disorders should be done for patients with unexplained thrombocytosis as they are at high risk of thrombotic complications.
- Ninety percent of patients with essential thrombocytosis have either JAK2, calreticulin or MPL mutation.
- Patients with essential thrombocytosis who have a thrombotic episode normally require lifelong anticoagulation.
KEYWORDS
Veno-occlusive disease, myeloproliferative disorders, Budd-Chiari syndrome, essential thrombocytosis, malignant hematology
CASE DESCRIPTION
A 43-year-old woman with no significant medical history presented with worsening abdominal distension and bloating for 3 weeks. She denied any alcohol use or recent changes in medications. Laboratory investigations revealed a slightly elevated white blood cell count (WBC) of 15,700 u/l with neutrophilic predominance, haemoglobin (Hb) 14.6 g/dl, and a platelet count of 449,000 u/l. A comprehensive metabolic panel showed deranged liver function tests (LFT): AST 61 (reference range 7–37) U/l, ALT 51 (reference range 10–49) U/l, ALP 322 (reference range 32–108) U/l, and total bilirubin 2.6 mg/dl. The patient underwent a computed tomography (CT) scan of the abdomen/pelvis and was found to have moderate-sized generalized abdominopelvic ascites in the absence of liver cirrhosis or hepatosplenomegaly, with diffusely decreased hepatic parenchymal density and periportal oedema. Ultrasound-guided paracentesis was performed with the removal of 5 litres of straw-coloured transudative fluid. No bacterial growth was seen on cultures and no malignant cells were seen on cytology. Further laboratory work revealed an elevated CA-125 level, so to rule out malignancy, the patient underwent a colonoscopy, which was normal. A pelvic sonogram revealed normal uterine contours and bilateral ovaries. She was discharged on spironolactone and furosemide for further investigation as an outpatient. Two weeks later when she was seen in the gastroenterology clinic, large volume ascites had reaccumulated and she was referred to the emergency department for paracentesis. Due to concern of veno-occlusive disease, a magnetic resonance venogram (MRV) of the liver was done which revealed narrowing of the intrahepatic and suprahepatic inferior vena cava (IVC) with a non-occlusive thrombus consistent with Budd-Chiari syndrome (BCS) (Fig. 1).
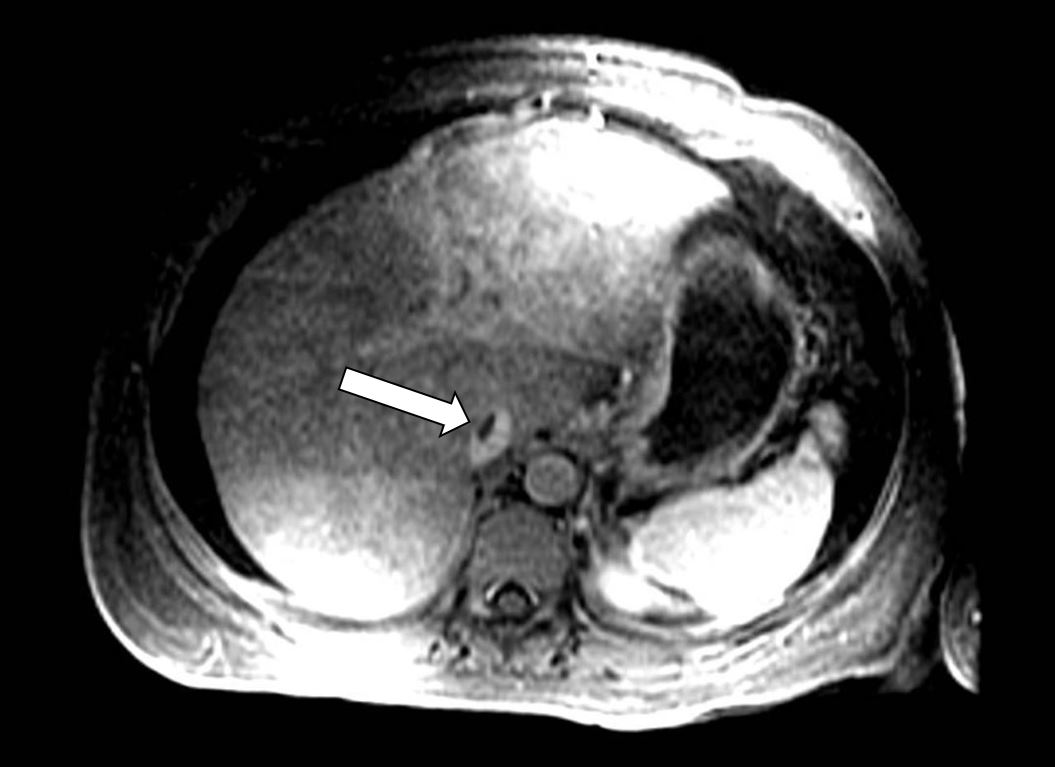
Figure 1 (click to enlarge)
Figure 1. Magnetic resonance venogram with intravenous contrast demonstrating a non-occlusive thrombus within the intrahepatic inferior vena cava (white arrow)
A heparin drip was initiated for anticoagulation and interventional radiology was consulted for possible mechanical thrombolysis, but the procedure was not performed due to the chronic nature of thrombosis. A hypercoagulable work-up was performed evaluating for Factor V Leiden deficiency, antithrombin III mutation, protein C and S deficiency, and antiphospholipid antibody syndrome (APAS) with anticardiolipin antibodies and anti-beta-2 glycoprotein antibody, all of which returned normal. On review of old records, the patient was found to have had a brief period of isolated thrombocytosis 2 years previously with a platelet count ranging from 700,000 u/l to 1,000,000 u/l (Fig. 2).
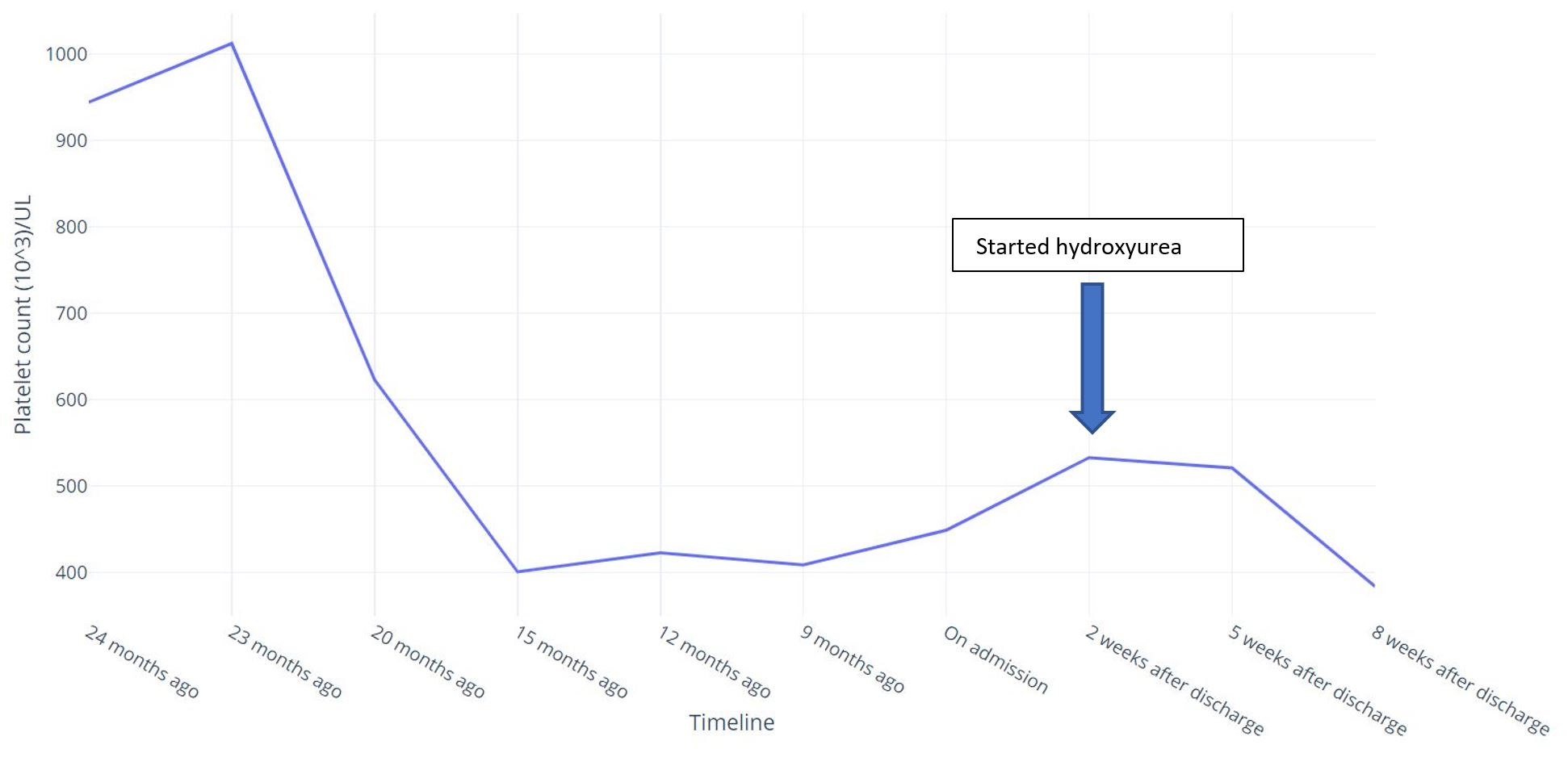
Figure 2 (click to enlarge)
Figure 2. Trend of the patient’s platelet count
Iron studies done at that time showed a normal ferritin level. JAK2 kinase mutation analysis and calreticulin mutation analysis were also negative. Thereafter her platelet count normalized spontaneously (Fig. 2). Although her platelet count was normal on admission, she was tested for MPL mutation as a possible cause of essential thrombocytosis (ET), which returned positive. She was discharged on apixaban for anticoagulation, and on follow-up with haematology after 2 weeks, repeat blood work showed mild thrombocytosis of 533,000 u/l and she started on myelosuppressive therapy with hydroxyurea 500 mg daily. Her platelet count dropped to within normal limits soon after initiation of therapy. Her ascites also improved with the use of diuretics and the intervals between repeat paracentesis gradually lengthened.
DISCUSSIONMyeloproliferative neoplasms (MPNs) are the most common underlying cause of BCS, occurring in approximately 50% of such patients [1]. In cases where there is no pre-established cause of thrombosis, other hypercoagulable disorders like paroxysmal nocturnal haemoglobinuria, protein C and S deficiency, and APAS also need to be ruled out [2]. Age-appropriate cancer screening is also recommended. Our patient had an elevated CA-125 level, which can be seen in patients with transudative ascites. Colonoscopy and pelvic ultrasound ruled out any malignancies. Essential thrombocythemia (ET) is an MPN characterized by excessive production of clonal platelets, increasing the risk of thrombotic events. Approximately 50% of patients with ET are diagnosed incidentally where thrombocytosis is discovered on routine blood work [2]. In females, it is important to consider iron deficiency as a possible culprit for thrombocytosis, although this is usually accompanied by concurrent anaemia [2]. Our patient, however, had normal ferritin and normal Hb. About 60% of patients with ET have JAK2 mutation, while 20–25% can have calreticulin mutation. MPL mutation is rarer, only seen in about 5% of patients, while 10–15% have ‘triple-negative’ ET where none of these driver mutations are detected [3]. As these patients are at a high risk of thrombosis, it remains imperative to follow patients with unexplained thrombocytosis and complete the diagnostic work-up. Our patient was lost to follow-up after JAK2 and calreticulin mutation assays returned normal and her platelet count fell into the high normal range. In such cases, the platelet count should be checked periodically, and a bone marrow biopsy should be done which can help confirm the diagnosis of an MPN [2].
The main goal of therapy in ET revolves around prevention and/or recurrence of thrombosis [1, 2]. Cytoreductive therapy is indicated in patients with hydroxyurea typically being the first choice [2, 4]. The data for concurrent use of low-dose aspirin is more convincing for polycythemia vera than for ET, although most experts favour its use [2]. Hydroxyurea is generally well tolerated, but patients can develop adverse effects of neutropenia or dermal ulcers [5]. In patients who develop catastrophic thrombotic complications like BCS or superior vena cava syndrome, lifelong anticoagulation with coumadin, or a direct oral anticoagulant, is warranted [2]. In patients with acute BCS, in addition to anticoagulation, early therapy with thrombolysis, both mechanical and pharmacological, has shown good results, however, the data for chronic thrombosis is less clear, as was the case in our patient essential
. In refractory cases, transjugular intrahepatic portosystemic shunt (TIPS) is an alternative for patients with worsening liver function and is often used as a bridge for a liver transplant, particularly in the paediatric population [4].
CONCLUSION
Clinicians should have a high suspicion for a ET in patients presenting with unexplained thrombocytosis. A complete work-up should be done even if the platelet count transiently decreases to normal range, and these patients should be referred to a haematologist. Early recognition and initiation of myelosuppressive treatment decreases the chances of thrombotic complications. However, if a thrombotic episode does arise, patients should be started on lifelong anticoagulation.