ABSTRACT
Anti-signal recognition particle (SRP) myopathy is a rare idiopathic inflammatory myositis that usually affects middle-aged women and is characterized by rapidly progressive proximal and symmetrical muscle weakness, elevated creatine kinase levels, severe necrotizing immune-mediated myopathy, presence of anti-SRP autoantibodies and poor response to steroid therapy. We report the case of a previously independent geriatric patient presenting with slow onset of proximal paraparesis, myalgia and severe gait impairment. Steroid and azathioprine treatment resulted in laboratory improvement and pain relief, but poor muscle strengthening. The atypical presentation delayed the correct diagnosis.
LEARNING POINTS
- Anti-signal recognition particle (SRP) myopathy is a rare idiopathic inflammatory myositis which can occur in the elderly.
- Atypical onset of the disease can hamper the diagnosis.
- SRP myopathy should be considered in the differential diagnosis of similar entities.
KEYWORDS
Signal recognition particle; myositis; anti-SRP antibodies; idiopathic inflammatory myopathies.
INTRODUCTION
Anti-signal recognition particle (anti-SRP) autoantibodies are found in 4-6% of idiopathic inflammatory myopathy (IIM) patients[1–3]. SRP is a polypeptide complex, involved in the translocation of recently synthesized proteins into the endoplasmic reticulum membrane[4]. Anti-SRP autoantibodies may be directed at any of the components of the SRP complex[2].
Despite the rarity of the disorder, all authors describe it as a severe necrotizing immune-mediated myopathy that usually affects middle-aged people and is characterized by rapidly progressive proximal and symmetrical muscle weakness, with severe disability occurring within months (sometimes accompanied by dysphagia), elevated creatine kinase (CK) levels (often extremely elevated), no recognizable seasonal pattern, and poor response to steroids or other traditional immunosuppressive therapies[1,4,5]. Histopathologically, it is defined by a necrotizing myopathy with little or no primary inflammation, weak expression of major histocompatibility complex class I (MHC-I), and the presence of particular patterns of complement C5b-9/membrane attack complex deposition[1,3,4]. Unlike antisynthetase syndrome, anti-SRP patients usually do not present with pulmonary fibrosis, arthritis, skin rash, fever or Raynaud's phenomenon, nor do they have an elevated risk for malignancies[3]. CK level seems to be an excellent indicator of the severity of myolysis in a given patient[4], useful for the follow-up of individuals, but it is a poor marker of disease activity at the population level.
CASE REPORT
An 88-year-old white woman, previously independent, presented to the emergency department (ED) with severe proximal paraparesis, myalgia (upper and lower extremities), anorexia, and paresthesias of the hands and feet. The muscle weakness had a slow onset over a year, with a significant rapid worsening (requiring the use of crutches), accompanied by myalgia in the previous 3 weeks, with no trauma history. The patient had a history of two hospital admissions in the previous year, when she had presented with rhabdomyolysis: the first admission was associated with pneumonia and dehydration, with CK levels of 2038 U/l (normal range <145 U/l), while the second occurred together with an episode of tachydysrhythmia with CK levels of 2209 U/l. In the second admission her statin drug was discontinued.
Examination showed symmetrical proximal paraparesis, grade 1/5 (Medical Research Council scale), with no weakness of the distal segments (grade 5/5) or the upper limbs. Myalgia was aggravated by muscle body palpation, especially of the quadriceps, gastrocnemius and deltoids. The patient did not present other relevant symptoms like fever, skin rash, arthralgias/arthritis, dysphagia to solids or liquids, dyspnoea or genitourinary symptoms.
Laboratory results showed elevated CK 4418 U/l, myoglobin 5075 ng/ml (normal range 9–82 ng/ml), CK-MB 402.4 ng/ml (normal range <3.6 ng/ml), troponin I 0.12 ng/ml (normal range 0–0.2 ng/ml), aldolase 54.9 U/l (normal range <7.6 U/l), lactate dehydrogenase (LDH) 1041 U/l (normal range <247 U/l), erythrocyte sedimentation rate (ESR) 41 mm/h (normal range 1–20 mm/h), discrete elevation of the hepatic enzymes and no renal impairment.
Electromyography and nerve conduction study showed signs of muscle fibre damage in the proximal muscles of the upper and lower extremities, associated with irritable myopathy. A biopsy sample of the deltoid muscle revealed an immune-mediated necrotizing myopathy with fibre diameter variability (few hypertrophic fibres, more frequent atrophic fibres, numerous necrotic fibres), no primary inflammation, increased nicotinamide adenine dinucleotide hydride tetrazolium reductase (NADH-TR) activity and little MHC-I activity (Fig. 1a–c). The autoimmunity study revealed a high titre of antibodies against SRP (by immunoblot and immunodot) and modest titres of anti-double-stranded deoxyribonucleic acid (anti-ds-DNA) autoantibodies and antinuclear antibody (ANA). The nailfold capillaroscopy was not suggestive of dermatomyositis/polymyositis (PM/DM) but revealed mild disorganization of the capillary architecture with multiple twisted capillaries and neovascularization.
The patient was treated with high-dose oral corticosteroids (48 mg methylprednisolone) and azathioprine (starting dose 50 mg) and physical rehabilitation was initiated. After 2 weeks of treatment, the patient showed minimal improvement in muscle strength (grade 2/5) but significant pain relief, with almost complete absence of myalgia and good laboratory response, with CK levels of 1623 U/l, LDH 703 U/l and ESR 13 mm/h. After 1 month of treatment, muscle strength had not improved further but CK levels had normalized (123 U/l). At this point, the patient developed a urinary tract infection, started antibiotics but progressed to urosepsis and died in hospital.
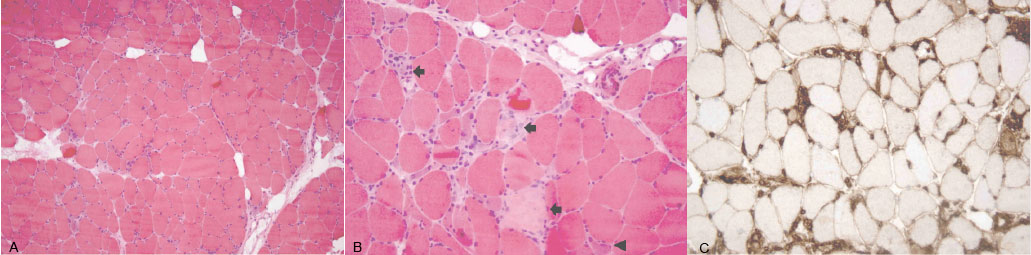
Figure 1 (click to enlarge)
Figure 1. (a) Abnormal muscle with fibre diameter variability and absence of inflammatory infiltrate (haematoxylin and eosin, magnification: 100×)
(b) Necrotic fibres at different stages (arrows) and regenerating basophilic fibres (arrowhead) (haematoxylin and eosin, magnification: 200×)
(c) Major histocompatibility complex class I (MHC-1) expression in apparently normal fibre sarcolemma (immunohistochemistry for MHC-1, magnification: 200×)
DISCUSSION
Although the primary characteristics of anti-SRP myopathy are fairly uniform, the disease shows substantial clinical heterogeneity, which makes recognition of the unusual pathological features extremely important for diagnosis and timely treatment. To our knowledge this paper reports the oldest patient yet described with anti-SRP myopathy as symptoms usually present in middle-aged people, more frequently in women[1,3].
Clinically, our patient presented with slow onset disease which was apparent during her previous admissions to the hospital with high CK levels. This slow onset might be associated with a worse neurological outcome than the rapidly progressive forms, which could explain why there was so little muscle strength improvement, despite normalization of CK levels (good for controlling the time course of the disease in each patient)[1]. However, this was not proved since our patient died 2 months after starting treatment. The slow onset of clinical muscle weakness delayed diagnosis as there were no other signs or symptoms to guide the medical investigation towards an immune-mediated myopathy until the patient presented with rapid progression associated with myalgia.
Complete or partial resistance to steroids is a common feature of anti-SRP myopathy[3,4], and for that reason our patient's initial therapy was a combination of steroids and azathioprine[5]. Other options have been proposed, but no combination has proven better than the others in terms of outcome.
In conclusion, anti-SRP myopathy patients may have polymorphic presentations, such as mild muscle weakness associated with elevated CK levels and, therefore, the correct diagnosis may not be considered promptly. The possibility of this condition should be suggested by other clinical presentations, such as myalgia associated with markedly raised CK levels, necrotizing myopathy with no primary inflammation and poor response to steroid treatment.