ABSTRACT
Primary adrenal lymphoma (PAL) is a very rare type of non-Hodgkin’s lymphoma (NHL). Herein, we report a case of NHL of both adrenal glands in a 69-year-old man. The patient was admitted because of a 1-month history of B symptoms and symptomatic hypotension. Biochemical analysis showed normocytic normochromic anaemia, hyponatraemia, hyperkalaemia, and elevated lactate dehydrogenase, C-reactive protein and D-dimers. A computed tomography scan revealed bilateral enlargement of the adrenal glands. There was no evidence of endocrine adrenal dysfunction. The mass in the right adrenal gland was biopsied and histopathology identified a diffuse large B-cell lymphoma of the activated B-cell subtype. A positron emission tomography 18F-fluorodeoxyglucose scan showed intensive hypermetabolic lesions involving both adrenal glands, as well as other locations, with higher uptake in the adrenal glands. Taken together, these findings suggested the diagnosis of PAL. The patient responded favourably to debulking therapy and is currently undergoing chemotherapy.
LEARNING POINTS
- Primary adrenal lymphoma is a rare condition presenting with unspecific symptoms; diagnosis requires histopathological confirmation.
- Adrenal function must be evaluated to rule out insufficiency.
- Positron emission tomography may reveal hitherto unsuspected extension of disease and should be performed where available.
KEYWORDS
Primary adrenal lymphoma, adrenal mass, positron emission tomography
CASE DESCRIPTIONS
A 69-year-old male patient with a 1-month history of chills, asthenia, anorexia, dry cough, precordial pain and symptomatic hypotension, was referred to our hospital by his primary care physician. His symptoms were exacerbated by consuming food, and he also reported night sweats, weight loss (5% in 1 month), nausea and sporadic non-bilious vomiting. He denied fever, loss of consciousness, pain in other locations, dyspnoea, changes in bowel movements, or urinary complaints. His medical history was relevant for type 2 diabetes mellitus, hypertension and dyslipidaemia.
On presentation, his physical examination was remarkable only for mild hypotension (114/76 mmHg). Initial laboratory findings revealed normocytic normochromic anaemia (haemoglobin 11.7 g/dl, normal range (NR) 12–15), hyponatraemia (Na+ 135 mmol/l, NR 136–145), hyperkalaemia (K+ 5.9 mmol/l, NR 3.5–5.1) and elevated lactate dehydrogenase (LDH) (277 U/l, NR <247 U/l), C-reactive protein (10.9 mg/dl, NR <0.5 mg/dl) and D-dimers (2845 ng/ml, NR <150). A thoracoabdominal-pelvic contrast-enhanced computed tomography (CT) scan was performed, revealing bilateral enlargement of the adrenal glands, measuring 55×54×73 mm on the right and 62×52×76 mm on the left (Fig. 1).
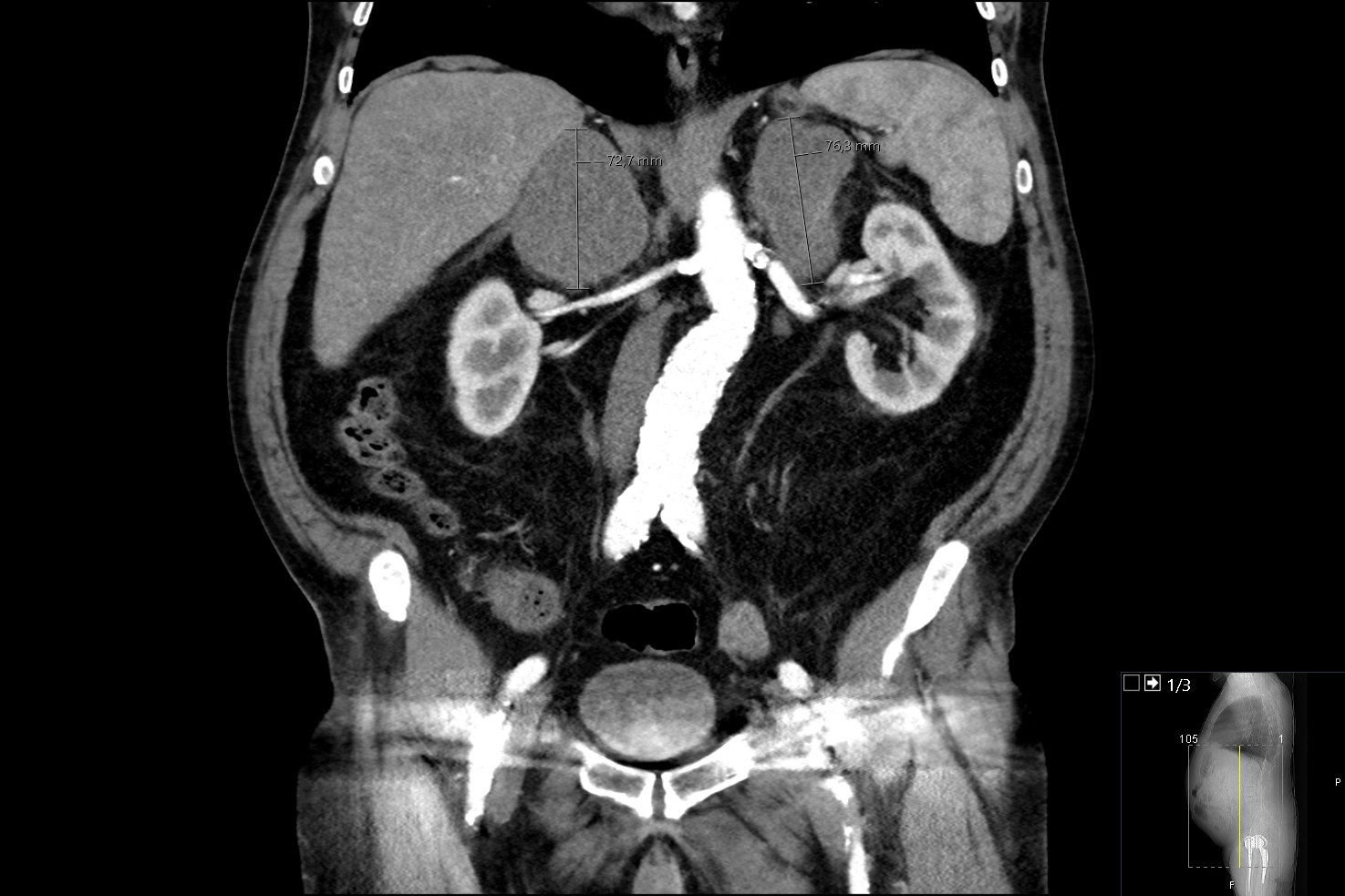
Figure 1 (click to enlarge)
Figure 1. Contrast-enhanced computed tomography (coronal view) of the abdomen and pelvis showing bilateral enlargement of the adrenal glands
everal abdominal ganglion formations were also observed, the larger ones at the proximity of the right adrenal mass, causing significant extrinsic compression of the inferior vena cava. Pulmonary embolism was ruled out.
The patient was admitted to the Internal Medicine ward for further investigation. During his stay, night fever of 38ºC was observed, with good response to paracetamol. Additional laboratory investigation excluded endocrine adrenal dysfunction (adrenocorticotrophic hormone 34 pg/ml, NR 9–52; serum cortisol 16 μg/dl, NR 5–25; plasma aldosterone 81.1 pg/ml, NR 40–310; aldosterone-to-renin ratio 0.42, NR <25; dehydroepiandrosterone SO4 1.29 μg/ml, NR 0.3–1.8; androstenedione 1.4 ng/ml, NR 0.6–3.7; urinary cortisol 48 μg/24 h, NR 10–80; urinary metanephrine 8.5 μg/24 h, NR 30–261; and urinary normetanephrine 241.3 μg/24 h, NR 50–521). Interferon γ assay was negative, as was serology for hepatitis B, hepatitis C and HIV.
Considering the most likely diagnosis of primary adrenal neoplasm, a CT-guided biopsy of the right adrenal gland was then performed, and histopathology revealed diffuse large B-cell lymphoma (DLBCL) of the activated B-cell subtype (Melan A–, CD3–, CD10–, CD20+, BCL-2+, BCL-6+, Ki67+, MUM1+). These findings, along with CT imaging, were consistent with the diagnosis of primary adrenal lymphoma (PAL).
The patient also underwent a positron emission tomography 18F-fluorodeoxyglucose scan (PET-FDG) which showed intensive hypermetabolic lesions involving both adrenal glands, the para-aortic lymph nodes, prostate, right testicle, both lungs, right spinalis muscle, and T9 vertebra (Fig. 2). There was no evidence of liver, spleen, kidney, pancreas or supra-diaphragm lymph node involvement, and the standard uptake value (SUV) was higher in the adrenal glands than other locations.
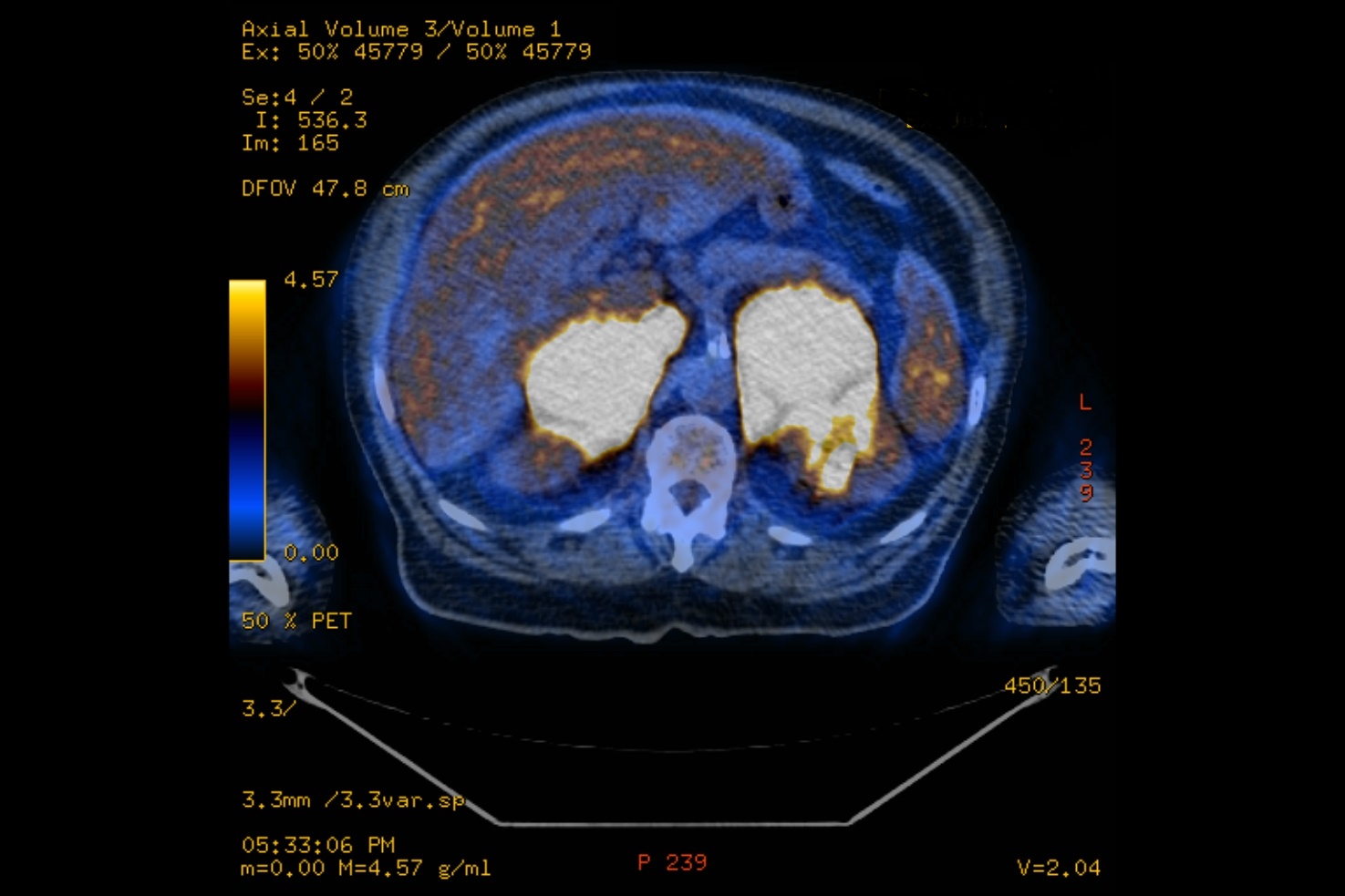
Figure 2 (click to enlarge)
Figure 2. Positron emission tomography 18F-fluorodeoxyglucose scan showing intensely hypermetabolic adrenal masses
Haematology-oncology consultation was obtained, and the patient was started on debulking therapy with 60 mg oral prednisolone daily, with resolution of his symptoms on the third day of treatment. After additional cardiology consultation, he was proposed for chemotherapy with cyclophosphamide, etoposide, vincristine and prednisolone (CEOP).
The patient is currently on the fourth cycle of chemotherapy, with no complications having been observed.
DISCUSSION
PAL has been defined as a histologically proven lymphoma involving one or both adrenal glands, in the absence of a previous history of lymphoma, and with predominant adrenal involvement when there are more organs or lymph nodes affected [1].
PAL is a rare but aggressive disease, with the largest review having found 187 cases [1], and further case series only slightly increasing this number [2, 3]. To the best of our knowledge, only two other cases have been reported from Portugal [4, 5]. Most patients were male, older than 60 years of age, and presented with B symptoms [1–3]. Anorexia, nausea and vomiting were reported less frequently [1–3]. The most common biochemical abnormality was elevated LDH, and DLBCL the most frequent histology [1–3].
Adrenal insufficiency was reported inconsistently, ranging from 11.4% to 61% of patients [1–3]. This may be due to lack of access to adequate testing in some locations [2], as well as the retrospective nature of the studies [1–3].
Most patients presented with bilateral rather than unilateral PAL [1–3], and with multifocal rather than exclusive adrenal disease [2, 3]. Of particular interest, PET-FDG was found to characterize extension of disease more accurately than other imaging methods [3]. Also, SUV was found to be higher in the adrenal glands than in other locations, which assisted in establishing the diagnosis of PAL [3].
Prognosis was significantly improved by combination chemotherapy, especially if the regimen included rituximab [1–3].
This case report is remarkable for describing the complex diagnostic and management challenge of PAL. CT-guided biopsy was essential in establishing the diagnosis and should be performed whenever non-invasive studies are inconclusive. PET-FDG revealed greater extension and metabolic activity of disease than previously suspected. This led to the decision to start debulking therapy in the hospital setting, and to withhold treatment with rituximab owing to the large risk of tumour lysis syndrome. Although we found no evidence of adrenal insufficiency in this case, we believe the patient’s hypotension may be explained by the tumoral mass compressing the inferior vena cava.